The
three mitotic protein kinases Aurora-A, B and C are complementary
enzymes that regulate multiple mitotic events. To do so, the different
kinases must be locally activated, and the control of their activity is
tightly regulated in time and space during mitosis. For instance,
Aurora-A is first active at the centrosomes, then on microtubules at the
spindle pole, Aurora-B is active in the nucleus, then at chromosome
kinetochores and later one at the midbody. Aurora kinase activity is
regulated in space and time by locally binding to regulators. Aurora
kinases must bind to protein partners to be activated. Aurora-A for
instance binds to targeting protein for Xenopus kinesin-like protein 2
(TPX2) and is activated at the spindle pole, Aurora-B and Aurora-C to
INner CENtromer Protein (INCENP) and is activated on the chromosomes.
These activations go through an autophosphorylation of a threonine
residue in the T-loop of the kinase. Other protein partners are using
different mechanisms to activate Auroras. These allow activation of the
kinase at different time and location in the cell. This review is an
up-to-date list of regulators of Aurora kinases. The subcellular
localization of these regulators explains the presence of an active
Aurora kinase. It also explains the changes in the localizations of the
Aurora kinases activity observed during cell cycle progression in
mitosis. Aurora kinases have been recently reported to be involved in
nonmitotic events, and the identity of their activators in these events
must be searched.
During the process of division, the cell goes through two
main phases such as interphase and mitosis that is followed by the
physical separation of the two daughter cells. During interphase, the
cell duplicates its contents that will be segregated during mitosis to
generate two daughter cells. The whole process lasts about 24 h in the
case of human cells during which mitosis takes only 1 h. This short
phase is highly regulated by phosphorylation and dephosphorylation
reactions [1].
Among the key protein kinases involved, there are cyclin-dependent
kinase 1 (CDK1), polo-like kinase 1(Plk1), NIMA-related kinase 2 (Nek2)
and the Aurora kinases (Aurora-A, B and C) [2]. The three mitotic protein kinases Aurora-A, B and C are complementary enzymes that regulate multiple mitotic events [3].
During mitosis, the cell segregates its two centrosomes that
migrate around the nucleus to reach opposite position. The nuclear
membrane breaks down, and the chromatin starts to condense to form
chromosomes. Microtubules nucleate from both the centrosomes and the
chromosomes to form a bipolar spindle [4].
The force exerted by the spindle microtubules contributes to the
alignment of chromosomes on the metaphase plate. This event is
immediately followed by the separation of each pair of sister chromatids
and the beginning of their migration to the two opposite poles of the
cell. The last part of the migration is driven by the central spindle,
assembled at the future site of cell division. A constriction ring is
assembled around the cell at the exact same location that will
contribute to the separation of two volumes corresponding to the two
daughter cells [5]. The physical separation of the two cells, abscission, will occur later on during the following interphase.
Aurora-A localizes at both the centrosomes and the spindle
poles. The kinase activity is required for microtubule nucleation during
bipolar spindle assembly and during central spindle formation [6].
Aurora-B is part of the Chromosome Passenger Complex (CPC), it
localizes at chromosome kinetochores from prophase to metaphase and at
the midbody from anaphase to telophase [7].
Its kinase activity is responsible for the massive phosphorylation of
the Ser10 of histone H3 in mitosis. Aurora-B corrects the wrong
attachments of microtubules to kinetochores during prometaphase [8].
During exit from mitosis, Aurora-B is required for cytokinesis.
Aurora-C that is mainly involved in meiosis can replace Aurora-B during
mitosis [9].
In vitro, the three kinases share the same substrates, such as histone H3. In vivo,
to fulfill their function, the three kinases are differentially
localized and also locally activated, and the control of their activity
is then tightly regulated in time and space during mitosis.
For
instance, Aurora-A is initially active in the cytoplasm and at the
centrosomes in the end of G2, then on microtubules at the spindle pole
during prometaphase.
Aurora-B is active in the nucleus by the end of G2,
then at chromosome kinetochores in prometaphase and later at the
midbody during anaphase and telophase. To achieve such regulation in
space and time Aurora kinases are locally activated by binding to
activators.
2. Bipolar spindle assembly
2.1. Aurora-A and TPX2
Targeting protein for Xenopus kinesin-like protein 2
(TPX2) is the best-characterized Aurora-Aactivator. It is a 100-kDa
protein expressed from G1/S transition to cytokinesis and then rapidly
degraded [10, 11]. TPX2 was first identified as a binding partner of the plus end-directed Xenopus kinesin-like protein 2 (Xklp2) [12]. TPX2 helps localizing Xklp2 to the spindle pole in prometaphase and metaphase [13, 14].
In interphase, from S to G2, TPX2 localizes to the nucleus
where it is sequestered by importin alpha. In mitosis, TPX2 is released
from the importin by RanGTP in the vicinity of the spindle. RanGTP
produced by the chromosome protein RCC1 (RanGEF) is localized as a
gradient around the chromosomes. It is only when the nuclear membrane
breaks down that TPX2 can reach the centrosome to bind Aurora-A, to
activate and re-localize Aurora-A protein on microtubules at the spindle
pole [15–17].
The binding of TPX2 to Aurora-A induces a conformational
change in the kinase in a way that the phosphorylated threonine of the
activation loop (T288 in human) is better protected from phosphatase
activity (PP1 in particular) [18].
Phosphorylation of T288 or binding to TPX2 triggers activation of
Aurora-A activity independently, but both the events are synergistic [19].
Most importantly, because binding of TPX2 to Aurora-A changes the
conformation of the kinase, it modifies the affinity of the substrates
for the kinase as well as the affinity of kinase inhibitors [20].
2.2. Aurora-A and CEP192
Centrosomal protein of 192 kDa (Cep192 ) was named after a proteomic analysis of the centrosome composition [21]. Cep192 is a protein involved in centrosome maturation and bipolar spindle assembly [22]. These functions correspond to those described for Aurora-A [23, 24], and indeed, Cep192 activates Aurora-A at the centrosome to control mitotic spindle assembly [25].
The mechanism by which Cep192 activates Aurora-A is different from
TPX2. Cep192 is a scaffold protein that brings together two molecules of
Aurora-A. Within the dimer, each Aurora-A molecule phosphorylates its
neighbor on T288, leading to the complete activation of the kinase. This
activation takes place at the centrosome early in mitosis. Aurora-A is
then presumably released from Cep192 to bind to TPX2 and to move on the
spindle poles [25].
2.3. Aurora-A and Maskin/TACC3
Maskin is a Xenopus laevis protein that got its
name by the fact that it regulates RNA translation. Maskin links the
5′cap and the 3′UTR of the mRNA, creating a closed structure that cannot
be translated [26]. During Xenopus laevis oocyte maturation, phosphorylation of Maskin by Aurora-A is required for the control of sequential mRNA translation [27].
Maskin is not only a substrate of Aurora-A, but it is also an activator
of its kinase activity as binding of Maskin to Aurora-A induces a
sevenfold stimulation of its kinase activity [27]. Whether Maskin affects the phosphorylation of threonine in the activation loop of the kinase has not been investigated yet.
The human homologue of Maskin is transforming acidic coiled-coil containing protein 3 (TACC3) and D-TACC in Drosophila melanogaster. Phosphorylation of D-TACC or TACC3 by Aurora-A is required for its centrosome localization [28], microtubule nucleation during bipolar spindle assembly [29–31] and during central spindle assembly [32]. Conversely, the activation of Aurora-A by TACC3 or D-TACC has not been demonstrated yet.
2.4. Aurora-B and INCENP
INner CENtromer Protein (INCENP) participates to the
chromosome passenger complex (CPC) together with Aurora-B, Survivin and
Borealin [33]. The complex controls multiple events during mitosis: from chromosome condensation and segregation to cytokinesis [34]. Aurora-B is carrying the kinase catalytic activity of the CPC, while INCENP is the activator of the kinase.
Binding of INCENP to Aurora-B is essential to the function of the kinase such as chromosome segregation and cytokinesis [35, 36].
Just like binding to TPX2 triggers activation of Aurora-A through
autophosphorylation of T288 in its activation loop, binding to INCENP
triggers activation of Aurora-B through autophosphorylation of T232 in
its activation loop [37, 38].
Both the kinases and the modes of activation are so closed that a
single amino acid change (G198 to N) transforms the activator of
Aurora-A from TPX2 to INCENP. The demonstration has been made in Xenopus [39] and human [40].
In term of evolution, it is striking to note that Drosophila melanogaster genome do not contain any gene coding for a TPX2. Ssp1/Mei-38 would be the closest TPX2-related protein in Drosophila.
Ssp1/Mei-38 possesses a microtubule-binding domain but lacks the
Aurora-A-binding domain, indicating that it cannot activate Aurora-A [41].
3. G2/M transition
3.1. Aurora-A and Ajuba
Ajuba means curiosity in Urdu, an Indian dialect.
Ajuba is a LIM domain-containing protein that serves as a scaffold to
build numerous protein complexes. The LIM domain is a Zinc finger
structure [42].
Ajuba was first reported to bind to Aurora-A at the centrosome in late
G2 and to trigger the kinase activation through autophosphorylation of
T288. Ajuba would then participate to the activation of Aurora-A and the
commitment to mitosis (Figure 1) [43]
. It was also suggested that Ajuba interacts with the N-terminal domain
of Aurora-A to release its inhibitory binding to the C-terminal
catalytic domain of the kinase [43].
Aurora-A activation by Ajuba would be a two-step mechanism, binding to
the N-terminal domain of the kinase and triggering autophosphorylation
of T288 [44]. The activation of Aurora-A by Ajuba has not been observed In Xenopus laevis [45]. In Drosophila melanogaster, although Ajuba does not activate Aurora-A, the protein is necessary to maintain Aurora-A at the centrosome [46].
3.2. Aurora-A and Nucleophosmin
Nucleophosmin (NPM) is a nucleolar protein involved in
multiple functions: histone chaperoning, ribosome biogenesis, mitotic
spindle assembly, genome stability, apoptosis and cancer [47]. Like Aurora-A, NPM localizes to the centrosome where it is required for centrosome duplication [48]. Depletion of NPM leads to the formation of disorganized spindles, a phenotype observed after Aurora-A depletion [49].
NPM is also a strong interactor of Aurora-A, and both proteins interact
at the centrosome late in G2. Binding of NPM triggers a phosphorylation
event on the kinase. The active Aurora-A, already phosphorylated on
T288, undergoes a second autophosphorylation on serine 89 which induces a
very strong stimulation of its kinase activity [50].
This stimulation is required at the centrosome in particular for the
phosphorylation of S353 on the phosphatase CDC25B involved in the
activation of CDK1/cylin B for G2/M transition. Surprisingly, the
stimulation of Aurora-A by NPM is not required for the phosphorylation
of T210 that activates PLK1 in the end of G2 [50] (Figure 1).
3.3. Aurora-A and Bora
Bora was identified in a genetic screen setup to search for mutations affecting the development of Drosophila melanogaster external sensory (ES) organs [51]. The gene was named Bora for Aurora-A Borealis because the phenotypes of Bora and Aurora-A mutants were similar. Bora binds to Aurora-A in vitro, is phosphorylated but also activates the kinase in Drosophila and human. In vitro,
Bora can activate Aurora-A in the presence of PP1 (seven- to
eightfold), suggesting that the mechanism used by Bora might be
identical to TPX2 although it was not demonstrated that Bora triggers
autophosphorylation of Aurora-A on threonine in the kinase activation
loop [51].
However, when expressed at physiological levels, Bora does not
co-immunoprecipitate with Aurora-A, and on the contrary, it
immunoprecipitates with PLK1. Furthermore, depletion of Bora does not
affect phosphorylation of T288 [52].
Eventually, it was demonstrated that Bora binds to the Polo-Box Domain
of Plk1 (PBD) to relieve the auto-inhibition of PBD and to expose the
T210 of the activation loop to Plk1-activating kinase. Aurora-A then
binds to Bora, gets activated and activates Plk1 by phosphorylating T210
(Figure 1) [52, 53]. This activation of Plk1 by Aurora-A through the interaction with Bora occurs in G2.
3.4. Aurora-A and AlBp1
AIBp1 (AIK binding protein, AIK stands for
Aurora/Ipl1-related kinase) is thus an Aurora-A binding protein but also
a hNinein binding protein. Depletion of AIBp1 gives phenotypes typical
of Aurora-A: bipolar mitotic spindle defects [54]. Binding of AIBp1 to Aurora-A increases its kinase activity in vitro [54]. In vivo expression of AIBp1 increases T288 phosphorylation on Aurora-A, whereas its depletion decreases T210 phosphorylation on Plk1 [55]. These data are reminiscent of the effect of Bora on both Aurora-A and Plk1 [52, 53]. It was then proposed that AIBp1 plays the same role as Bora but in a hNinein signaling pathway.
Figure 1.
The
three kinases CDK1/cyclinB, Plk1 and Aurora-A phosphorylate substrates
required the G2/M transition. This scheme shows the pathways used by
Aurora-A to activate Plk1 and CDK1/cyclinB.
4. Actin network
4.1. Aurora-A and HEF-1/NEDD9/Cas-L
Human enhancer of filamentation 1 (HEF1) or neural precursor
cell expressed, developmentally down-regulated 9 (NEDD9) or
Crk-associated substrate related, lymphocyte-type (Cas-L) is a
scaffolding protein that localizes to focal adhesions in interphase
cells and to the mitotic spindle in M-phase. It participates in
integrin-dependent signaling processes, such as cell attachments, cell
migration and cell survival [56].
Cells depleted with HEF-1 show a decrease in T288 phosphorylation of
Aurora-A, indicating that HEF-1 is required for activation of Aurora-A
kinase in vivo. In vitro both proteins directly
interact, and when increasing levels of HEF-1 are added to Aurora-A, an
increase of T288 phosphorylation and its kinase activity are observed [57].
Interaction of HEF-1 with Aurora-A occurs in G2, during which the
activation of Aurora-A by HEF-1 induces phosphorylation of HEF-1 and
inhibition of the interaction. Interaction of HEF-1 with Aurora-A plays a
critical role in primary cilia disassembly upon reentry in the cell
cycle after Go arrest. In this particular case, HEF-1-activated Aurora-A
phosphorylates and activates HDAC6, which in turn deacetylates the
tubulin that is sufficient to provoke cilia resorption [58].
The activation of Aurora-A in the process of cilia disassembly is also
dependent on Ca2+ and calmodulin (CaM) that are required for Aurora-A to
bind to its activators [59].
4.2. Aurora-A and PAK-1
p21-Activated protein kinase-1 (PAK-1) regulates cell motility and morphology [60, 61]
and is involved in focal adhesion disassembly through the PAK-PIX-GIT
complex, PIX is a Rac GTP exchange factor and GIT is a G-protein-coupled
receptor kinase-interacting protein [62].
This complex is also active at the centrosome, and when Pak1 is
activated it dissociâtes from the PIX-GIT to phosphorylate and activate
Aurora-A [63].
The activation goes through phosphorylation of the T288 in the
activation loop, but there was also a phosphorylation of S342 in the C
terminal end of the kinase. Although T288 is known to be an activation
site, S342 is rather known to inhibit Aurora-A kinase activity when
phosphorylated. This has been shown in Xenopus laevis where the phosphorylation of S349 (human S342) downregulates Aurora-A in vitro [64] and in vivo during oocyte maturation between Metaphase I and Metaphase II of meiosis [65].
The same data have been reported in human where phosphorylation of S342
was observed in G2 upon DNA damage to inhibit Aurora-A, avoiding
mitosis entry in the presence of lesions [66]. This discrepancy has not been solved yet.
4.3. Aurora-A and ILK
Integrin-linked kinase (ILK), like PKA-1, is a protein
kinase involved in cell adhesion, and the kinase links the extracellular
matrix to the actin cytoskeleton [67, 68].
ILK has also been observed in centrosome where it associates with
TACC3/Ch-TOG, and its kinase activity is required for Aurora-A
interaction with TACC3 [69]. ILK acts upstream of Aurora-A that in turn phosphorylates TACC3 on S558, to control microtubule nucleation [70, 71]. How ILK controls Aurora-A activity toward TACC3 is unknown.
4.4. Aurora-A and Arpc1b
Arpc1b is a component of the seven-subunit protein Arp2/3 complex involved in new actin filament nucleation and polymerization [72].
Arpc1b localizes on centrosome in G2 and interacts with Aurora-A only
if Arpc1b has been previously phosphorylated on T21 by Pak-1. Arpc1b is
also a substrate of Aurora-A, and the kinase phosphorylates wild-type
Arpc1b but not the T21A mutant. This phosphorylation by Aurora-A is
required for the interaction of Arpc1b with Arpc2 [73]. On the other hand, Arpc1b is an activator of Aurora-A in vitro and in vivo,
and binding to Arpc1b triggers T288 autophosphorylation just like TPX2
does. Depletion of Arpc1b leads to a decrease on T288 as well as a
decrease of its activity toward substrates such as PLK1 or histone H3 [73].
5. Ubiquitylation
Ubiquitination corresponds to a posttranslational
modification (PTM) of proteins during which 76 amino-acid peptides are
covalently linked to a protein, usually on lysine residues. It requires a
multistep reaction: it needs 1) an E1 enzyme that will activate the
ubiquitin, then 2) an E2 enzyme that will conjugate the ubiquitin and
finally 3) an E3-ligase that will catalyze the transfer of ubiquitin
peptide on the protein substrate [74].
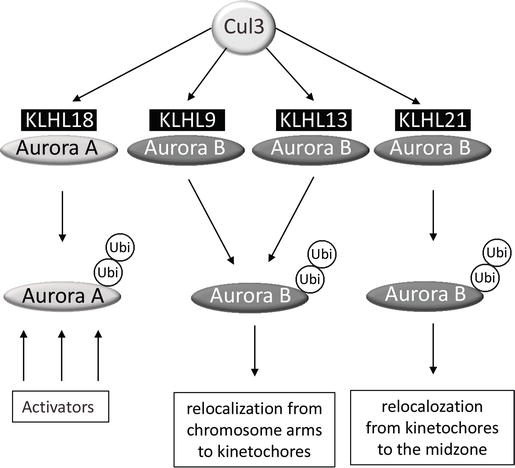
5.1. Aurora-A and CUL3-KLHL18
The multiprotein complex E3 ubiquitin ligases of the
cullin-RING-type ubiquitin ligase family include eight members in human.
In the case of Cul3, the broad-complex, tramtrack and bric-a-brac (BTB)
domain-containing proteins like KLHL18 (Kelch-like) serve to recognize the ubiquitin substrate [75]. There are about 200 BTB proteins in human with various functions not all being Cul3 adaptors [76].
Cul3 and KLHL18 localize at the centrosome in late G2, and the
depletion of each of the protein provokes a delay in the G2/M transition
that has been attributed to a default in Aurora-A phosphorylation on
T288 and consequently a default in the kinase activation at the
centrosome [77].
The activation of Aurora-A by Cul3-KLHL18 involved a nonproteasomal
ubiquitination of the kinase; however, the activation is not a direct
effect of ubiquitination (Figure 2).
Although the mechanism of activation is not fully understood, the
hypothesis is that ubiquitination of Aurora-A could facilitate the
interaction of the kinase with its activators in late G2, such as Ajuba
or Cep192 for instance (Figure 1).
5.2. Aurora-B and CUL3-BTB proteins
Aurora-B unlike Aurora-A binds to three different BTB
proteins KLHL9, KLHL13 and KLHL21, and all three substrate adaptors
participate to Cul3 complexes that ubiquitinate Aurora-B in vivo and in vitro [78, 79].
Like for Aurora-A, the ubiquitination of Aurora-B does not lead to any
degradation of the kinase, although depletion of Cul3, KLHL9 and KLHL13
mimics depletion of the 26S proteasome [78].
On the contrary, ubiquitination by Cul3-KLHL9, −KLHL13 or KLHL21
regulates the kinase localization during mitosis. In the absence of
KLHL9 or KLHL13, instead of moving to the kinetochore region, Aurora-B
remains on chromosome arms during prometaphase/metaphase [78]. In the absence of KLHL21 instead of moving to the midzone, Aurora-B remains on anaphase chromosomes (Figure 2).
In this last case, it is the whole CPC complex with Aurora-B, INCENP,
Survivin and Borealin that remains on anaphase chromosomes [79].
Although KLHL9, KLHL13 and KLHL21 are substrate adaptors,
their localizations do not really fit to their function. KLHL9 and
KLHL13 have not been found on chromosomes for instance. On the other
hand, KLHL21 does localize to the midzone where it could bring Cul3 to
ubiquitinate Aurora-B, but even in this case KLHL21 should be at the
kinetochore to ubiquitinate Aurora-B to target it to the midzone. The
proposed hypothesis to explain this discrepancy is the high turnover of
Aurora-B on its localization.
.
Figure 2.
Cul3-dependent
ubiquitination of Aurora kinases. Aurora-A is targeted by KLHL18, while
Aurora-B by KLHL9, KLHL13 and KLHL21. Ubiquitination of Aurora-B
localizes the protein, while ubiquitination of Aurora-A stimulates
binding of the activators.
5.3. Aurora-A, Aurora-B and FBXW7
FBXW7 is an F-box protein participating to Skp, Cul1, F-box
containing complex (SCF), a multiprotein E3 ubiquitin ligase complex
that ubiquitinates proteins to be degraded by the proteasome. There are
69 F-box proteins coded by the human genome. Like KLHL proteins for
Cul3, F-box proteins target the ubiquitin ligase complex to Cul1
substrates. FBXW7 binds to Aurora-A and Aurora-B and participates to
their ubiquitination in vivo and in vitro [80, 81]. Depletions of FBXW7 stabilize both protein kinases levels in vivo, indicating that the FBXW7-dependent ubiquitination leads to the degradation of the kinases by the proteasome [81, 82].
F-box proteins usually recognize phosphorylated proteins, and FBXW7 for
instance binds to Aurora-A previously phosphorylated by GSK3β on S245
and S387 [81] (Figure 3). Phosphorylation sites involved in Aurora-B/FBXW7 have not been identified, and the kinase involved is not known.
Figure 3.
Cul1-dependent
ubiquitination of Aurora kinases. Both Aurora-A and Aurora-B are
targeted by FBXW7 F-box for ubiquitination. Aurora-A must be previously
phosphorylated by GSK3β, while the kinase phosphorylating Aurora-B is
unknown. Ubiquitination triggers degradation.
5.4. Aurora-A, Aurora-B and Cdh1
Cdc20-homologue 1 (Cdh1) is an activator and
substrate adaptor of the E3 Ubiquitin ligase APC/C (Anaphase Promoting
Complex/cyclostome). Cdh1 recognizes proteins containing a D-box
(destruction box) (motif…RxxL…) and a KEN-box (motif …KEN…). All three
Aurora kinases (A, B and C) contain a D-box in the carboxy end of the
protein [83]. Both Aurora-A and B are ubiquitinated and degraded in a D-box-dependent manner [83, 84] although there are conflicting reports regarding Aurora-B [85].
Whether Aurora-C is degraded through its D-box remains also an open
question. The Cdh1-dependent degradation of Aurora-A was demonstrated in
both Xenopus and human [86, 87].
A new sequence required for the Cdh1-dependent degradation was
discovered in Aurora-A from different species that was absent in
Aurora-B and C. This sequence in the NH2 terminal end of the protein was
named simultaneously A-box (for only in Aurora-A) [88] and DAD-box (for D-box-activating domain) (Figure 4) [89].
Interestingly, the A-box of Aurora-A contains a Serine at position 51
that when phosphorylated stabilizes the protein by inhibiting the
functionality of the A-box [86, 90].
Figure 4.
CDH1-dependent
ubiquitination of Aurora kinases by APC/C. Both Aurora-A and Aurora-B
are targeted by APC/C to be degraded. The ubiquitination required the
presence A- and D-boxes in Aurora-A as well as the unphosphorylated
state of S51 and the presence A- and D- and KEN-boxes in Aurora-B.
When tested in Xenopus laevis extracts Aurora-B was
not found to be degraded in a Cdh1-dependent manner, it does contain a
D-box but no A-Box, and only a chimera protein Aurora-A/Aurora-B
containing Aurora-A A-box and Aurora-B D-box could be degraded in the
extract [88, 89]. However, study in human finally revealed that Aurora-B contains also a functional D-box recognized by Cdh1 [84].
Interestingly, the same authors report that the KEN-box in Aurora-B is
required for Cdh1-dependent degradation. The discovery of a functional
A-box in Aurora-B was more surprising, and since the box is not only
present in Aurora-A, the name DAD-box seems now more adequate than A-box
to name it. Finally, Aurora-B needs three functional boxes to be
degraded in a Cdh1-dependent manner from the NH2 to the COOH end: a
KEN-Box (KEN), an A-box (QRVL) and a D-box (RxxL) [85].
6. Sumoylation
Sumoylation resembles ubiquitination, and it is a
posttranslational modification corresponding to a covalent attachment of
a one or several SUMO proteins (100 amino acids) to a substrate. SUMOs
stands for small ubiquitin-related modifiers, and there are now about
ten different ubiquitin-like modifiers including ubiquitin and SUMO [91].
Like for ubiquitination, the sites of sumoylation are lysine residues,
and the modification occurs also in a three-step reaction by E1, E2 and
E3 ligases.
Both Aurora-A and Aurora-B are sumoylated in vivo on a lysine residue located in the sequence …IHDRIKPEN… conserved in all Aurora kinases [92, 93].
Aurora-A is sumoylated on K249, and expression of the
Aurora-A mutant K249R that cannot be sumoylated affects spindle assembly
and potentiates the oncogenic property of the kinase [93].
Aurora-B is sumoylated on lysine 207, and expression of the Aurora-B
mutant K207R affects chromosome segregation and cytokinesis [92].
Interestingly enough, sumoylation of Aurora-A or Aurora-B does not
affect the kinase activity in vitro indicating that sumoylation is
probably playing required for the localization of the protein or for
protein-protein interaction. The exact function of Aurora sumoylation
remains to be found.
7. Poly(ADP-ribosyl)ation
Poly(ADP-ribosyl)ation (PARylation) is a covalent
modification of protein that can be catalyzed by 16 different
Poly(ADP-ribose) polymerases (PARP) that attach multiple ADP-ribose
units on substrate proteins by hydrolyzing NAD+. The reverse reaction is
insured by Poly(ADP-ribose) glycohydrolase (PARG) (three isoforms) and
several ADP ribose hydrolases [94, 95]. Only Aurora-B is PARylated in vivo, and interestingly enough, the modification occurs in the presence of DNA damage [96].
PARylation of Aurora-B leads to a decrease of Serine 10 histone H3
phosphorylation, indicating a loss of Aurora-B kinase activity. The
kinase directly interacts with PARP-1 and PARP-2, and both enzymes can
PARylate Aurora-B and inhibit its activity [96].
8. Phosphorylation
Aurora kinases belong to a family of protein kinases that
need to be phosphorylated on a threonine residue in its T-loop to be
active [97]. Aurora-A must be phosphorylated on T288, Aurora-B on T232 and Aurora-C on T198 and T202 [37, 98, 99].
These phosphorylations are autophosphorylation events that occur in the
presence of an activator such as TPX2 for Aurora-A or INCENP for
Aurora-B and C [16, 99].
Aurora-A has two other levels of regulation controlled by
phosphorylation. The kinase activity can be upregulated by
autophosphorylation of S89 in the presence of nucleophosmin, as
described above [50]. Aurora-A can also be downregulated by phosphorylation of S342 [64]. In vivo,
phosphorylation of S342 occurs in the presence of DNA damages during G2
and depends on the activation of the checkpoint kinase Chk1 [66, 100]. Is Aurora-A a direct target of Chk1 or the target of a kinase downstream of Chk1 remains an open question.
Since Aurora kinases are phosphorylated, they are obviously
targeted by phosphatases. T288 in Aurora-A and T232 in Aurora-B for
instance are dephosphorylated and inactivated by type 1 protein
phosphatase [101, 102]. T288 is also dephosphorylated by PPP6 that specifically target Aurora-A when bound to TPX2 [103]. These dephosphorylations inactivated Aurora kinase activity.
As discussed above, Aurora-A degradation by APC/C-CDH1
depends on the presence of an A-box in the kinase and phosphorylation of
S51 within the human A-box inhibits this degradation process [64].
Although the kinase responsible for this phosphorylation remains to be
identified, the phosphatase PP2A insures its dephosphorylation [104].
9. Conclusion
Since their discovery, Aurora kinases have become priority targets for the development of inhibitors for cancer treatments [105].
But their regulation takes multiple forms, adding difficulties in
developing the efficient drugs targeting the kinases. This review tends
to report a nonexhaustive list of posttranslational modifications (PTMs)
affecting the functions of the kinases. These PTMs can be used as
biomarkers, like the phosphorylation of T288 in Aurora-A frequently used
to measure the kinase activity in vivo, and this test is currently questioned [106].
More interestingly, these PTMs can be used to design original
inhibitory strategies different from those targeting the kinase active
site. The binding of TPX2 to Aurora-A for instance has been targeted to
search for Aurora-A inhibitor [107].
This kind of approach targeting PTMs offers broad prospects for
specific inhibition of Aurora kinases. Many new inhibitors should then
be discovered in the coming years.
Acknowledgments
We wish to apologize to the authors who were not quoted in
this review. Work in the laboratory is supported by grants from the
“Ligue Nationale Contre le Cancer” (LNCC, équipe labelisée 2014–2016 and
2017), the CNRS and the University of Rennes 1. APD venus was supported
by “La Fondation Rennes 1.” LV, APD are fellow of the LNCC and Région
Bretagne, while OG was supported by “La Fondation pour la Recherche
Médicale” (FRM).




Inga kommentarer:
Skicka en kommentar