
Löysin yllöättäen asettaessani vanhoma abstrakteja kansioon yhden abstraktin, joka käsitteli sirtuiini-inhibiettoria.
SEIFERT Tina (2014). Modulationg Sirtuin Activity. Design, synthesis and evaluation of Sirtuin 2 inhibitors.
ISBN 978-91-628-9249-4.
Katson mitä lähteitä samalla löydän:
2009
https://pubs.acs.org/doi/abs/10.1021/jm8014298
Novel Cambinol Analogs as Sirtuin Inhibitors: Synthesis, Biological Evaluation, and Rationalization of Activity
The tenovins and cambinol are two classes of sirtuin inhibitor that
exhibit antitumor activity in preclinical models. This report describes
modifications to the core structure of cambinol, in particular by
incorporation of substitutents at the N1-position, which lead to
increased potency and modified selectivity. These improvements have been
rationalized using molecular modeling techniques. The expected
functional selectivity in cells was also observed for both a SIRT1 and a
SIRT2 selective analog.2012
Synthesis and Evaluation of Substituted Chroman-4-one and Chromone Derivatives as Sirtuin 2-Selective Inhibitors
† Department of Chemistry and
Molecular Biology, Medicinal Chemistry, University of
Gothenburg, SE-412 96 Göteborg, Sweden
§ Department of Medicinal Chemistry,
R&I iMed, AstraZeneca R&D, SE-431
83 Mölndal, Sweden
⊥School
of Pharmacy and ∥Department of Neurology, Institute of Clinical
Medicine, University of Eastern Finland, P.O. Box 1627,
70211 Kuopio, Finland
J. Med. Chem., 2012, 55 (16), pp 7104–7113
DOI: 10.1021/jm3005288
Publication Date (Web): July 2, 2012
Sirtuins (SIRTs) compose class III of lysine deacetylases (KDACs). There
are seven conserved human isoforms (SIRT1–SIRT7) with different
subcellular locations.(1)
The enzymes catalyze the reversible deacetylation of lysine residues
both on histones (H1, H3, H4) and nonhistone proteins, e.g., p53, p65,
PGC-1α, PPARγ, FOXO, NFκB, and α-tubulin.(2) The deacetylation reaction requires nicotinamide adenine dinucleotide (NAD+) as cosubstrate, and results in the formation of the deacetylated protein substrate, O-acetyl-ADP-ribose, and nicotinamide, which is the endogenous inhibitor of the sirtuins.(3-5)
In addition to the deacetylation, also mono-ADP-ribosyl transferase
activity and removal of long-chain fatty acyl groups from lysine
residues have been reported for SIRT6.(6-8) Mono-ADP-ribosyl transferase activity is the only effect observed for SIRT4.(9) Recently, Du et al. discovered lysine demalonylation and desuccinylation activities of SIRT5,(10)
which later was followed by reports by Zhao and co-workers regarding
deglutarylation from lysine residues as a function of SIRT5.(11)
Because
of the broad spectrum of substrates, SIRTs have been implicated as
regulators in a range of physiological processes, including metabolism,
cell survival and apoptosis, gene expression, and DNA repair.(12)
Therefore, the enzymes have been proposed to be involved in pathologies
such as inflammation and aging-associated diseases, e.g., cancer,
diabetes, and neurodegeneration (e.g., Alzheimer’s, Huntington’s, and
Parkinson’s disease).(13, 14) The potent SIRT1 inhibitor selisistat (36 (Ex-527), Chart 2) reduces Huntington’s disease pathology(15) and has been in phase II clinical studies.(16)
SIRT2,
which is the focus of the present study, is predominantly located in
the cytoplasm but is enriched in the nucleus during mitosis.(17)
Beside the deacetylation of histone H4, the enzyme is also involved in
the deacetylation of nonhistone substrates such as α-tubulin, FOXO, p65,
p300, and p53.(18) Hence, the enzyme is proposed to be involved in the regulation of the cell cycle.(19)
SIRT2 is highly expressed in the brain,(18)
and inhibition appears to be neuroprotective as two SIRT2 selective
inhibitors have been shown to counteract progression of Huntington’s(20, 21) and Parkinson’s disease.(22)
Regarding its role in oncogenesis, there are contradictory reports in
the literature whether SIRT2 is a tumor suppressor or promoter.(23-25) Down-regulation of SIRT2 reduced the cell proliferation in glioma cells,(26) HeLa cells,(27) and liver(28) and pancreatic carcinomas.(29) Inhibition of SIRT2 by selective inhibitors such as 30 (AGK-2, Chart 2) and a 10,11-dihydro-5H-dibenz[b,f]azepine derivative have been shown to induce apoptosis in C6 glioma cells(26) and MCF-7 breast cancer cells,(30)
respectively. Other small-molecule SIRT2 inhibitors have shown to
reduce cancer proliferation via the increase in p53 acetylation in
nonsmall-cell lung cancer cells (A549 and H1299).(31) Hence, SIRT2 has been considered to be an interesting target for cancer drug development.
Recently, we showed that trisubstituted 2-alkyl-chroman-4-ones can serve as selective SIRT2 inhibitors with IC50 values in the low micromolar range.(32) Two of the most potent inhibitors are shown in Chart 1.

Chart 1. SIRT2 Selective Inhibitors Based on the Chroman-4-one Scaffold
The
structure–activity relationship (SAR) study revealed that
electron-withdrawing groups on the aromatic ring of the bicycle, the
carbonyl group, as well as an alkyl side chain in the 2-position are
crucial for potent inhibitors. The SAR study also disclosed an
exceptionally close relationship between the presence of all features
mentioned above and the inhibitor potency as even minor modifications
resulted in a severe loss of activity. Analysis of the individual
enantiomers of 1 showed that the stereoisomers had only small differences in inhibitory activities, with (S)-1 being slightly more potent (Chart 1).
However,
the high lipophilicity of the published chroman-4-ones limits their use
in more advanced biological in vivo and in vitro tests. Herein, we
report chroman-4-one analogues based on lead compounds 1 and 2
as potential SIRT2 inhibitors with increased hydrophilicity. The
hydrophilicity was increased by the introduction of heterofunctional
groups such as terminal hydroxyl, carboxylic acid, ester, and amide
moieties in the alkyl side chain in the 2-position of 1. The phenyl ring in 2
was replaced with aromatic and aliphatic heterocycles. We also propose a
binding mode of the chroman-4-ones based on a homology model of SIRT2.
It showed to be consistent with the SAR data of the investigated
compounds. Two potent inhibitors were chosen for further evaluation of
their effect on cancer cells.
Results and Discussion
Chemistry








The
key reaction in the synthesis of the biologically evaluated compounds
is the assembling of the chroman-4-one scaffold. The general synthetic
strategy for the formation of chroman-4-ones 6a–i is shown in Scheme 1
and involves the base-promoted aldol condensation between a substituted
2′-hydroxyacetophenone and an aldehyde, followed by an intramolecular oxa-Michael ring closure reaction. Commercially available alcohols (3a, 3d–g) were used as precursors for the desired aldehydes, whereas alcohols 3b and 3c
were synthesized via monoprotection of commercially available diols
using NaH and TBDMSCl according to a procedure reported by McDougal et
al.(33) The aldehydes (4a–g)
were obtained via Swern or Dess–Martin oxidation and could be directly
used in the next step without any further purification. For the ethylene
glycol based aldehyde 4d, the ordinary workup procedure
involving addition of water and EtOAc had to be changed to a nonaqueous
workup due to its high water solubility.
Scheme 1. General Synthetic Scheme towards the Substituted Chroman-4-ones 6a–ia
aReagents and conditions: (a) (i) (COCl)2, DMSO, THF, −78 °C, 30 min, (ii) appropriate alcohol, −78 °C, 30 min, (iii) Et3N, −78 °C → room temp, 15 min, or Dess–Martin periodinane, CH2Cl2, room temp; (b) appropriate 2′-hydroxyacetophenone, DIPA, EtOH, MW, 170 °C, 1–2 h; (c) Selectfluor, MeOH, MW, 150 °C, 30 min. bCommercially available hexanal (4h) was used.
The chroman-4-ones 5a–c and 6d–i
were synthesized in moderate to good yields by heating
2′-hydroxyacetophenones and the appropriate aldehydes in a microwave
reactor for 1 h in ethanol using N,N-diisopropylamine (DIPA) as base. The TBDMS-protection group of 5a–c was removed using electrophilic fluorine in a microwave-heated reaction,(34) providing the deprotected chroman-4-ones 6a–c in 16–78% yield over three steps. Removal of the silyl protecting group of 5a and 5b with the more commonly used tetrabutylammonium fluoride (TBAF) in THF surprisingly yielded the ring opened products 7a and 7b (Scheme 2). No formation of the ring-opened byproduct was observed for 5c. Treatment of the hydroxyl derivatives 6a and 6b
with TBAF also resulted in the formation of ring opened products. The
byproduct formation is likely to be attributed to the basicity of the
fluoride ion in organic solvents. In separate experiments, it was
however found that the byproduct was not formed when triethylamine was
used as base.
Scheme 2. Synthesis of Ring-Opened Derivatives Using TBAFa
aReagents and conditions: (a) TBAF, THF, room temp, overnight.
The ester analogues of 6a and 6b were prepared according to the synthetic route outlined in Scheme 3. δ-Valerolactone and γ-butyrolactone were ring-opened under basic conditions in MeOH. Alcohol oxidation provided aldehydes 8a and 8b, which were reacted with 3′-bromo-5′-chloro-2′-hydroxyacetophenone to afford 9a and 9b (Scheme 3).
Scheme 3. Synthesis of Ester Derivatives of 2,6,8-Trisubstituted Chroman-4-onesa
aReagents and conditions: (a) Et3N, MeOH, room temp, 18 h; (b) SO3·pyridine, Et3N, DMSO, room temp, 14 h, or (i) (COCl)2, DMSO, THF, −78 °C, 30 min, (ii) appropriate alcohol, −78 °C, 30 min, (iii) Et3N, −78 °C → room temp, 15 min; (c) 3′-bromo-5′-chloro-2′-hydroxyacetophenone, piperidine or DIPA, EtOH, 170 °C, 0.5–1 h.
As
esters are prone to hydrolyze in vivo, we also wanted to test the
corresponding carboxylic acid analogues for their SIRT2 inhibitory
potency, as well as amide and oxadiazole analogues, which are
hydrolytically more stable and also considered to be ester bioisosteres.
Chroman-4-ones 9a–b were believed to be good starting
points to access the carboxylic acids as well as the different amide
analogues. However, attempts to hydrolyze the ester functionality of 9b under basic conditions (LiOH or Me3SnOH)
were unsuccessful. Neither were attempts to obtain the amide analogues
by directly reacting 2-hydroxyacetophenones with δ-amido aldehydes,
obtained by ring opening of δ-valerolactone with amines followed by
oxidation of the obtained amido alcohols, successful.
Instead, oxidation of the primary alcohol of 6a and 6b by a Dess–Martin oxidation, followed by a Pinnick oxidation of the generated aldehydes, yielded the carboxylic acids 10a–b (Scheme 4). The acids were then successfully coupled with different primary and secondary amines using N,N′-carbonyldiimidazole (CDI) as coupling reagent to provide amides 11a–e in good to excellent yields (68–93%). Treatment of the CDI-activated acids 10a or 10b with acetamide oxime followed by heating provided the oxadiazoles 12a–b.
Scheme 4. Synthesis of Chroman-4-one-Based Carboxylic Acids 10a–b, Amide Analogues 11a–e, and Oxadiazoles 12a–ba
aReagents and conditions: (a) Dess–Martin periodinane, CH2Cl2, room temp, 0.75–1 h; (b) NaClO2, NaH2PO4·2H2O, amylene, H2O, THF, 0 °C → room temp; (c) (i) appropriate acid, CDI, CH2Cl2/DMF,
0 °C, 30 min, (ii) appropriate amine, 0 °C → room temp, 2–14 h; (d) (i)
appropriate acid, CDI, MeCN/DMF, room temp, 30 min, (ii) acetamide
oxime, 85 °C, 14–19 h.
To further explore the influence of the phenyl ring in lead compound 2 (Chart 1), pyridine rings (6e–g)
and morpholine, piperidine, or piperazine moieties were planned to be
incorporated. The latter chroman-4-ones were envisioned to be
synthesized in analogy to the standard route outlined in Scheme 1 starting from commercially available alcohols as precursors (Scheme 5).
However, neither Swern oxidation of 3-morpholinopropan-1-ol nor the use
of other oxidizing agents such as Dess–Martin periodinane, TEMPO, TPAP,
or CrO3(35) resulted in the desired aldehyde 13. Finally, 13 was obtained by conjugate addition of morpholine to acrolein (Scheme 5). Applying the standard procedure for approaching target compound 14a
was unsuccessful as well as attempts via preformation of the aldol
intermediate using of lithium diisopropylamide (LDA). Attempts to
approach the aliphatic heterocycles containing chroman-4-ones via a
substitution reaction of a terminal hydroxyl group failed due to the
unsuccessful reaction of the acetophenone and aldehyde 15. None of the approaches resulted in the formation of the desired products.
Scheme 5. Synthetic Attempts towards Chroman-4-ones 14a and 14ba
aReagents and conditions: (a) MeCN, MgSO4, room temp, overnight, 93% crude yield; (b) (i) (COCl)2, DMSO, THF, −78 °C, 30 min, (ii) 3-(tert-butyldimethylsilyl)oxy-1-propanol, −78 °C, 30 min, (iii) Et3N, −78 °C → room temp, 15 min, 97% crude yield; (c) 3′-bromo-5′-chloro-2′-hydroxyacetophenone, DIPA, EtOH, 170 °C, 1 h.
Eventually,
the prolongation of the spacer between the chroman-4-one scaffold and
the heterocycle to propylene enabled the synthesis of related analogues
of the phenethyl-substituted chroman-4-one 2. The synthetic pathway toward the derivatives is outlined in Scheme 6. Compounds 17a and 17b were finally prepared from the mesylated chroman-4-one 16 via a microwave-assisted substitution reaction using morpholine and piperidine.
Scheme 6. Synthesis of Chroman-4-ones 17a–b with Aliphatic Heterocyclic Rings in the 2-Positiona
aReagents and conditions: (a) MsCl, Et3N, CH2Cl2, 0 °C → room temp; (b) morpholine or piperidine, THF, MW, 120–150 °C, 1 h.
In
addition to the above-described monocyclic heterofunctionalities,
bicyclic groups were introduced to move the hydrogen-bonding groups
further away from the scaffold. Two different ring systems were chosen,
i.e., quinolin-6-yl and 3,4-dihydro-2(1H)-quinolinone-6-yl. The starting material 6-bromo-3,4-dihydro-2(1H)-quinolinone was prepared in analogy to the procedure reported by Tietze et al.(36) and Zaragoza et al.(37) Reaction of the bromo-substituted bicyclic systems with acetal-protected acrolein in a Heck reaction yielded 18a–b (Scheme 7).(38) Catalytic hydrogenation and deprotection of the acetal under acidic conditions furnished the desired aldehyde 19a (Scheme 7). Surprisingly, under the mild reducing conditions (H2-balloon, 10% Pd/C, room temp) chosen to reduce the aliphatic double bond in 18b also the quinoline moiety was reduced to yield the corresponding 1,2,3,4-tetrahydroquinoline. When Pd/C and 1,4-cyclohexadiene(39)
was used as reducing agent, only reduction of the aliphatic double bond
occurred, and after treatment with acid, the desired aldehyde (19b)
was obtained. The aldehydes were then reacted under standard conditions
with 3′-bromo-5′-chloro-2′-hydroxyacetophenone to yield 20a–b in moderate yields.
Scheme 7. Synthesis of Chroman-4-ones Containing Bicyclic Heterofunctional Groups in the Side Chain in the R2-Positiona
aReagents and conditions: (a) appropriate aryl bromide, acrolein diethyl acetal, Pd(OAc)2, KCl, K2CO3, TBAA, DMF, 90 °C, overnight; (b) H2,
10% Pd/C, MeOH, room temp, 3 h or 1,4-cyclohexadiene, 10% Pd/C, EtOH,
reflux, 4.5 h; (c) HCl (conc), acetone, reflux, 2–4 h; (d)
3′-bromo-5′-chloro-2′-hydroxyacetophenone, DIPA, EtOH, MW, 160–170 °C,
1.5–2 h.
The tetrasubstituted chromones 21–25 were synthesized as illustrated in Scheme 8. The monobrominated chroman-4-one 21 was obtained by reaction of 2 with CuBr2. Treatment of 21 with NaN3 in DMSO resulted in the formation of amine 22,(40) which was acetylated with acetyl chloride in pyridine to form 23. A SmI2-mediated
Reformatsky type reaction using tosyl cyanide as described earlier by
Ankner et al. was successfully applied to introduce a nitrile moiety in
the 3-position, and subsequent oxidation with DDQ in dioxane yielded
3-cyano-chromone 24.(41) Further reduction of the nitrile group by means of DIBAL-H furnished enaminone 22 in 66% yield.
Scheme 8. Synthesis towards Tetrasubstituted Chromone Derivativesa
aReagents and conditions. (a) CuBr2, CHCl3/EtOAc, 2 h, reflux; (b) NaN3, DMSO, 70 min, room temp; (c) AcCl, pyridine, room temp, overnight; (d) SmI2, KHMDS, TsCN, THF, −78 °C → room temp, cis:trans 29:71; (e) DDQ, dioxane, room temp, 12 h; (f) DIBAL-H, CH2Cl2, −78 °C, 3 h.
Molecular Modeling
Mode of Action of Sirtuins and Inhibitor Binding

Sirtuins
contain a conserved enzymatic core comprising a Rossmann fold domain
and a small domain containing a three-β-stranded zinc binding motif. The
Rossmann fold contains six parallel β-strands forming a central β-sheet
which is sandwiched between α-helices (number dependent on SIRT
isoform) on either side of the β-sheet.(42, 43) Between these domains the binding sites of NAD+ and the acetylated peptide substrate are located. The NAD+
binding site can be formally divided into the subpockets A (adenine
binding site), B (ribose binding site), and C (nicotinamide binding
site).(42) The binding of the peptide substrate and NAD+ is proposed to take place in a sequential manner.(44) Binding of the peptide substrate induces the cleft between the two domains to close, and upon NAD+ binding the cofactor binding loop gets ordered.(43, 45) The presence of an acetyl lysine peptide promotes a strained, productive NAD+
conformation, which is required for the deacetylation reaction to
proceed. This conformation positions the nicotinamide moiety of NAD+ in the C-pocket and brings the ribose ring in vicinity of the acetyl group in the peptide substrate.(42, 43, 46)
The C-pocket is the presumable binding site for potent small-molecular SIRT1/2 inhibitors (Chart 2) such as 30,(22) thieno[3,2-d]pyrimidine-6-carboxamides (31),(47) 3-arylideneindolin-2-ones (32),(48) salermide (33),(49) cambinol (34),(50) and splitomicin analogues (35)(51) and 36.(52) We therefore wanted to investigate whether this pocket could be a feasible binding site also for our compounds.
Chart 2. Structures of SIRT1/2 Inhibitors Known to Occupy the C-Pocket of the NAD+-Binding Site and Our Chroman-4-one-Based Inhibitor 6f
Homology Modeling of Human SIRT2

Four crystal structures of SIRT2 are currently available.(53-55) Two of these are apo-structures (PDB codes 1J8F and 3ZGO (3ZGO is a rerefined structure of the human SIRT2 apoenzyme 1J8F)),(53, 54) lacking both peptide substrate and NAD+ in their catalytic site. Their structures differ considerably compared to structures where peptide substrate and/or NAD+ are bound. Another SIRT2 structure in complex with ADP-ribose (ADPr) was recently solved (3ZGV).(54) ADPr is similar to NAD+ in structure; it binds in the NAD+
binding cleft and thereby induces the conformational change which
closes the active site crevice around ADPr in the Rossmann-fold domain.
The fourth structure (4L3O) is a complex of SIRT2 with inhibitor S2iL5, which is a macrocycle binding from the outside into the peptide binding channel.(55) Because the structure with ADPr (3ZGV) lacks a bound inhibitor and includes ADPr instead of NAD+ and the macrocycle in the latest solved structure (4L3O)
largely differs from the small molecular chroman-4-ones, we decided to
construct a homology model of SIRT2 based on a more suitable template.
Recently, crystal structures were published of both SIRT3 and Sir2Tm with (S)-36 (Chart 2) present in the C-pocket.(52) Also SIRT1 binding (S)-37 (Chart 2) has been crystallized.(56) The Sir2Tm structure (PDB code 4BV2) is a complex which includes NAD+, (S)-36,
and a peptide substrate, but as it has a rather low resolution (3.3 Å)
it was considered not to be the most suitable template for homology
modeling. The structures that are solved of SIRT3 with inhibitor (S)-36 (PDB 4BV3) and SIRT1 with (S)-37 (PDB 4I5I)
bound in the C-pocket seemed suitable as the size and shape of these
compounds are similar to the chroman-4-one-based inhibitors. Of the two
structures considered, the SIRT3/NAD+/(S)-36 structure (PDB 4BV3) binds NAD+
in a nonproductive mode, with the nicotinamide moiety rotated 180°
relative to the C-pocket and instead positioned in the peptide binding
channel. Like (S)-36 and (S)-37, the chroman-4-ones do not appear to be peptide substrate competitive inhibitors (data not shown), therefore the SIRT1/NAD+/(S)-37 complex (PDB code 4I5I) was chosen as a template for homology modeling.(56) Models were constructed that included a lysine residue from a Sir2-p53 peptide–NAD+ complex (2H4F)(46) together with NAD+ and inhibitor (4I5I). The homology modeling was performed using the MOE software (v. 2012.10, Chemical Computing Group Inc.: Montreal).
A multiple sequence alignment was performed in ClustalW (v. 2.1)(57) using the template (SIRT1 human, 4I5I (Q96EB6)),(56)
the main target (SIRT2 human, Q8IXJ6), and the human SIRT3 sequence
(Q9NTG7). The alignment was fine-tuned to improve the final homology
model (Figure S1, Supporting Information). A detailed description of the homology modeling procedure is given in the Supporting Information.
The force field used in the homology modeling in this study was
Amber12:EHF with R-Field solvation, as implemented in the MOE software.
During the construction of the homology model, inhibitor 6f was positioned in the C-pocket close to the location of 37 in the SIRT1 structure in order to achieve more information regarding the inhibitor–enzyme interactions. NAD+
was included in the modeling procedure with its geometry kept from the
SIRT1 structure, i.e., in its nonproductive mode. The lysine residue
from a Sir2-p53 peptide–NAD+ complex (2H4F) was included,(46) as were five structural water molecules from the SIRT1 structure (4I5I)
positioned within 5 Å from the inhibitor (W2, W5, W17, W74, and W78).
The selected inhibitor-induced SIRT2 homology model showed good
geometrical properties (see Ramanchandran plots in Figure S2 in the Supporting Information). In the resulting homology model the chroman-4-one scaffold of inhibitor 6f is buried in a hydrophobic and well-defined binding pocket. The carbonyl oxygen of 6f forms a hydrogen bonding interaction with a conserved water molecule W17 (Figure 1).(52, 54, 58)
There is also a halogen bonding interaction from the chloride in the
6-position to the backbone carbonyl of His187 with a distance of 3.7 Å
(Cl···O═) and an angle of 163.5° (C–Cl···O═), which is an acceptable
geometry.(59)
The bromide in the 8-position is located in a hydrophobic environment
surrounded by Leu103, Phe119, Leu138, and Phe190. In addition, Phe96 is
favorably positioned for π–π interaction with the halogen substituted
aromatic ring of 6f(60)
although this interaction is not geometrically optimal as the rings are
arranged in a shape which is in between a parallel displacement and a
face-to-edge arrangement (r = 5.4 Å, θ = 53.7°, ω = 35.1°, for definition see Figure S3 in Supporting Information).(61) The 2-(pyridin-3-yl)ethyl moiety is positioned in a rather narrow hydrophobic channel (Figure 1), which is directed toward the surrounding solution. The pyridine nitrogen of 6f
forms a hydrogen bond with Gln142 but could instead easily interact
with surrounding water molecules outside the enzyme (Figure 1a,b).
Figure 1. (a) Schematic view of the interactions between the chroman-4-one-based inhibitor 6f and the human SIRT2 homology model. The carbonyl oxygen of 6f interacts with a structural water molecule, which in turn interacts with a glutamine residue (Gln167) and with NAD+.
The bromide in the 8-position is buried in a hydrophobic environment
(Leu103, Phe119, Leu138, and Phe190) and the chloride in the 6-position
can form a halogen bonding interaction to the backbone carbonyl of
His187. The substituent in the 2-position is stretched through a
hydrophobic tunnel surrounded by Ile93, Pro94, Leu103, and Leu138, which
ends in the aqueous environment at the surface of the enzyme, where the
pyridyl nitrogen interacts with Gln142 via hydrogen bonding. (b) SIRT2
homology model with inhibitor 6f (magenta) present in the
C-pocket indicating the same interaction points as in (a). The carbonyl
group of the acetylated substrate peptide interacts with a hydroxyl
group in the ribose moiety of NAD+.
Evaluation of Inhibitory Activity



The
inhibitory effect of the synthesized chroman-4-one/chromones on the
activity of SIRT2 was evaluated using a fluorescence-based assay. Table 1 summarizes the results from the SIRT2 inhibition assay of the trisubstituted chroman-4-ones. For potent inhibitors, IC50
values were determined, and these compounds were also tested against
SIRT1 and SIRT3. In general, highly potent inhibitors showed to be
selective for the SIRT2 subtype (for SIRT1 and SIRT3 inhibition data,
see Table S3 in Supporting Information).
Table 1. Results from Evaluation of Trisubstituted Chroman-4-ones in a SIRT2 Inhibition Assay
Table a
SD, standard deviation (n = 3).
Table b
Inhibition at 200 μM inhibitor concentration. Suramin was used as reference compound in the assay.
Table c
IC50 (95% confidence interval). IC50
values were determined for compounds showing >80% inhibition of
SIRT2 at 200 μM concentration or compounds evaluated in the cell
proliferation assay.
Table d
n.d.= not determined.
Table e
The SIRTAinty assay was used for the determination.
The SIRT2 inhibition data for the tetrasubstituted chromones are summarized in Table 2. These chromone-based derivatives are moderately potent and selective SIRT2 inhibitors (Table 2).
Table 2. Results from Evaluation of the Tetrasubstituted Chromones in a SIRT2 Inhibition Assay
| no. | R3 | inhib (%)a,b | IC50 (μM)c,d |
|---|---|---|---|
| 22 | NH2 | 79 ± 1.5 | nd |
| 23 | NHAc | 81 ± 0.9 | 28.7 (21.4–38.5) |
| 24 | CN | 50 ± 1.9 | nd |
| 25 | 76 ± 1.9 | nd |
a
SD, standard deviation (n = 3).
b
Inhibition at 200 μM inhibitor concentration.
c
IC50 (95% confidence interval). IC50 value was determined for compounds showing >80% inhibition.
d
nd = not determined.
Potent
inhibitors were also tested for their inhibitory effect on members of
other classes of HDACs. The test confirmed that the compounds
exclusively inhibit the class III of lysine deacetylases (HDAC
inhibition <10 200="" at="" data="" div="" not="" shown="">
Physicochemical properties of this new series of chroman-4-one-/chromone-based inhibitors were calculated (Table S4, Supporting Information).
The results indicate that the structural modifications did lead to less
lipophilic compounds with improved physicochemical properties. As
illustrated in Table 3, the potent inhibitors of the new series have similar inhibitory activity as lead compounds 1 and 2,
however they exhibit more attractive physicochemical properties such as
decreased clogP and clogD-values as well as a larger PSA (Table 3). Among the synthesized compounds, derivatives 6f and 12a were chosen for evaluation of a potential antiproliferative effect (see below Figure 3). Although the methyl ester 9b is the most potent inhibitor and 9a is as active as 12a,
the methyl ester group is prone to hydrolysis in the cell-based assay,
yielding the biologically inactive carboxylic acids analogues 10a and 10b, therefore 9a and 9b were not further investigated.
Table 3. Data of Calculated Physicochemical Properties of Lead Compounds 1 and 2 and of 6f, 9a–b, and 12a from the New Series
| no. | IC50 (μM) | MW | ACDlogP | ACDlogD pH 7.4 | PSA (Å2) | HBDa | HBAb |
|---|---|---|---|---|---|---|---|
| 1 | 4.3 | 331.6 | 5.60 | 5.60 | 27.4 | 0 | 2 |
| 2 | 6.8 | 365.7 | 5.57 | 5.57 | 27.4 | 0 | 2 |
| 6f | 3.7 | 366.6 | 4.19 | 4.18 | 37.6 | 0 | 3 |
| 9a | 9.6 | 347.6 | 3.36 | 3.36 | 54.4 | 0 | 4 |
| 9b | 2.0 | 361.6 | 3.77 | 3.77 | 54.4 | 0 | 4 |
| 12a | 12.2 | 371.6 | 3.69 | 3.69 | 61.0 | 0 | 5 |
a
Number of hydrogen bond donors.
b
Number of hydrogen bond acceptors.
Structure–Activity Relationships

In
this study, we have focused on chroman-4-ones/chromones with increased
hydrophilicity. This was achieved by the introduction of aliphatic and
aromatic mono/bicyclic moieties with hydrogen-bonding groups in the
2-position of the chroman-4-ones as well as the implementation of
hydrogen bonding groups in the 3-position of the chromones. These
changes were done to improve the inhibition of SIRT2 but also to obtain
information about space limitations caused by the introduction of
bulkier groups.
The substituent in the
2-position is crucial for SIRT2 inhibition because 6-bromochroman-4-one
lacking a substituent in the 2-position does not show any inhibition
(data not shown), while the corresponding analogue 6h, which has a pentyl group in the 2-position, shows an inhibitory activity of 70% (Table 1). Replacement of the pentyl side chain in lead compound 1 with a more polar ethylene glycol side chain (6d) resulted in a significant decrease in activity (33% inhibition) as did the introduction of a terminal hydroxyl group (6a, 18% inhibition). However, by increasing the length of the linker between the OH-group and the scaffold (6b and 6c), some activity could be retained (Table 1). Still, 6c (67% inhibition) is less potent then the lead compound ((rac)-1, 88%). This indicates that highly polar side chains (6d)
and hydrogen bond donating groups are not favorable with a chain length
up to five atoms. This observation can be explained by the homology
model, which shows that five out of seven amino acids surrounding this
channel are hydrophobic (Ile93, Pro94, Leu103, Leu138, and Phe143,
Figure 1a and Figure S1 in the Supporting Information).
One of two hydrophilic amino acids (Asp170) is rotated away from the
substituent in the 2-position while the other (Gln142) is directed
toward the aqueous solution, which might explain the enhanced activity
with increasing length of the spacer for alcohols 6a–c. Replacement of the phenyl ring in 2 with a pyridyl moiety (6e–g) resulted in compounds with similar activity and improved solubility. The 3-pyridyl substituted chroman-4-one (6f) was with 86% inhibition most potent compared to the 2- and 4-pyridyl analogues 6e and 6g. Modeling results showed that the pyridyl moiety of 6f
has an optimal geometry to form a hydrogen bond with Gln142, which is
located at the end of the hydrophobic channel toward the aqueous
solution (Figure 1b). This hydrogen bonding interaction can also be observed for the ester functionality in 9a and 9b, the latter being one of the most potent inhibitors with an IC50
value of 2.0 μM. As earlier mentioned, esters are prone to hydrolysis
under physiological conditions and therefore also the corresponding
acids were investigated. However, the carboxylic acids 10a and 10b
were completely inactive, which could be attributed to carboxylate
formation at physiological pH. Negatively charged groups are unfavorable
in the lipophilic and narrow channel accommodating the R2-substituent. The replacement of the methyl ester in 9a–b with a methyl amide (11a and 11b) dramatically lowered the activity. Also replacement of the methyl amide with the bulkier iso-propyl (11c) or benzyl amides (11d) gave no increase in inhibitory potency. Only the dimethyl amide 11e was slightly more potent, with 53% inhibition compared to 39% for the monomethyl amide. The secondary amides (11a–d)
can act both as hydrogen bond accepting and donating group, and here it
seems as the presence of an NH-group decreases the activity. However,
all amide analogues are not as active as the methyl esters (9a–b).
An explanation for this could be that an NH-group is less favorable in
the lipophilic environment than the less hydrophilic single bonded
oxygen of the ester. This statement was supported in docking studies(62, 63) of 9b and 11b, where the O- and N-methyl groups are positioned in a small hydrophobic pocket rather than toward the aqueous solution (Figure 2).
Thereby, the single-bonded oxygen in the ester and the NH-moiety in the
amide are oriented toward the hydrophobic site, which makes the more
polar amide unfavorable compared to the ester. Compound 11e is still less active than 9b, which could be explained by the steric bulk of the dimethyl amide in the narrow tunnel.
Figure 2. Docked chroman-4-one analogues 9b (red) and 11b
(green) in the SIRT2 homology model. The green part of the surface is
hydrophobic while the purple is hydrophilic. The carbonyl oxygens are
forming hydrogen bonds with the glutamine residue Gln142, and the methyl
groups are positioned in a small hydrophobic pocket. The polar hydrogen
on the amide is pointing toward a hydrophobic region. Only the water
molecule interacting with the carbonyl group in the chroman-4-ones (W17)
is shown.
The oxadiazole derivative with an ethylene linker (12a, IC50
= 12.2 μM) was equipotent to the corresponding methyl ester.
Surprisingly, the bioisosteric replacement of the methyl ester moiety of
9b with an oxadiazole group (12b) resulted in lower potency. Docking studies showed that 12a can adopt a similar binding pose as the ester whereas the methyl group of the longer oxadiazole 12b did not fit in the binding channel. The morpholine- and piperidine-substituted analogues 17a and 17b
showed 17% and 40% inhibition, respectively. The morpholine and
piperidine moieties are rather bulky, and the extended spacer places the
groups further away from the scaffold and closer to the narrow part of
the channel. Beside this, piperidine and morpholine are charged at pH
7.4, which seems unfavorable in the hydrophobic binding pocket. The
chroman-4-one derivatives with the quinolinone and quinoline moieties (20a and b)
were moderate inhibitors with 59% and 56% inhibition, respectively.
These results are consistent with a previously published
indolyl-substituted derivative,(32)
which had 53% inhibitory activity. The rather narrow hydrophobic
channel accommodating the side chain seems to be large enough to
accommodate monocyclic ring systems rather than the large bicyclic
moieties. Interestingly, the 6-bromo-8-chloro-chroman-4-one derivative 6i (IC50 1.8 μM) showed twice the activity of racemic 1 (IC50
4.3 μM), which strengthens the hypothesis of a halogen bonding
interaction between the halide in the 6-position and the backbone
carbonyl. A bromide is a better halogen bonding group than chloride, and
therefore 6i was supposed to be more active than lead compound 1.
The chromones (Table 2)
with an additional substituent in the 3-position showed generally only
moderate inhibitory activity (50–81%). The acetamide substituted
phenethyl-chromone 23 was the best inhibitor, with 81% inhibition and an IC50
value of 29 μM. The introduction of the small heterofunctional side
chain on the flat ring system did not result in the desired increase in
potency via additional hydrogen bonding interactions.
Evaluation of Antiproliferative Properties



We have previously shown that SIRT1/2/3 pan-inhibitors(64) with a well-documented mechanism of sirtuin inhibition(45, 65)
can cause antiproliferative effects in MCF-7 breast cancer and A549
lung cancer cell lines. Literature reveals that also SIRT2 inhibitors
have been shown to have an antiproliferative effect in MCF-7 breast
cancer cells(30, 66) and A549 lung cancer cells.(31) We therefore wanted to study whether the novel compounds could achieve similar effects. Two potent inhibitors (6f, 12a)
with acceptable physicochemical profiles were chosen for testing in
cancer cell proliferation assays. These two human cancer cell lines were
exposed to increasing concentrations of 6f and 12a, and
the cell proliferation was measured using a sulforhodamine B assay. Both
compounds had a strong inhibitory effect on cancer cell growth (Figure 3). Compound 6f
showed a significant antiproliferative effect already at 10 μM
concentration, and under microscopic evaluation there were no living
cells visible after 48 h with higher concentrations (≥50 μM) of 6f (data not shown).
Figure
3. SIRT2 inhibitors reduce A549 (left) and MCF-7 (right) cancer cell
proliferation. The cells were treated with 0–100 μM of 6f and 12a.
Cell proliferation was determined by a sulforhodamine B assay. The
results are shown as mean ± SEM of two to three independent experiments.
The asterisks indicate significant differences (* P < 0.05, *** P < 0.001 when compared to controls).
SIRT2 is known to deacetylate α-tubulin.(67) To confirm the functionality of 6f and 12a
as SIRT2 inhibitors in a cellular environment, MCF-7 cells were treated
with these compounds and subjected to Western blot analysis of
α-tubulin acetylation levels. After 6 h, treatment with 100 μM 6f had produced a drastic increase in acetylated α-tubulin and lower concentrations of 6f had weaker effects to the same direction (Figure 4). After 18 h, all cells treated with 100 μM 6f had died and no sample could be obtained for Western blotting. Nevertheless, 6f
gave significant levels of inhibition of α-tubulin deacetylation at
lower concentrations at this time point. Treatments with 50 and 100 μM
of 12a decreased total α-tubulin after 18 h. When compared to
total α-tubulin, there was a clear, albeit nonsignificant trend toward
increased α-tubulin acetylation in these samples.
Figure
4. Effects of SIRT2 inhibitors on α-tubulin acetylation. MCF-7 cells
were treated for 6 h (left panel) or 18 h (right panel) with 40 nM
Trichostatin A plus indicated concentrations of 6f or 12a. The results are shown as mean ± SEM of three independent experiments. The asterisks indicate significant differences (* P < 0.05, *** P < 0.001 when compared to controls). The representative Western blots are shown below.
Flow cytometric cell cycle analysis was performed in order to examine the basis for the antiproliferative effects of 6f and 12a. Treatment of MCF-7 or A549 cells for 18 h with 100 μM 12a resulted in cell cycle arrest, as there was a significant increase in the fraction of cells in G1/G0 phase and a significant decrease in the fraction of cells in DNA synthesis phase (Figure 5). Furthermore, the fraction of A549 cells in G2 phase decreased. Treatment with 50 μM 6f also resulted in similar cell cycle arrest in A549 cells (significant) and MCF-7 cells (trend) (Figure 5). No apoptosis was observed in any of the samples (data not shown). The G1/G0
arrest is not necessarily resulting from increased acetylation of
α-tubulin but may result from some other SIRT2-mediated event, and
similar observations have been found in the literature.(31, 68-70)
Figure
5. Effects of SIRT2 inhibitors on A549 (left) and MCF-7 (right) cell
cycles. The cells were subjected to control treatment (0.5% DMSO) or
treatment with 6f (50 μM) or 12a (100 μM) for 18 h. Flow
cytometric analysis of DNA content was done after propidium iodide
staining. Percentage of cells in each phase of the cell cycle (G1/G0, S, and G2/M)
is indicated. The results are shown as mean ± SEM of two to four
independent experiments. The asterisks indicate significant differences
(* P < 0.05, ** P < 0.01, *** P < 0.001 when compared to controls).
Conclusion
A
series of chroman-4-ones carrying heterofunctional groups in the side
chain in the 2-position together with four tetrasubstituted chromones
were synthesized in good yields using efficient synthetic methods.
Compared to the previously published chroman-4-ones, calculations of the
physicochemical properties indicate that the new compounds show
improved pharmacokinetic properties. Analogues carrying hydrogen bond
accepting groups, e.g., pyridyl or ester moieties, were highly potent
and selective SIRT2 inhibitors with low micromolar IC50 values. Two compounds (6f and 12a)
were chosen for investigation of their effects on cell proliferation in
MCF-7 breast cancer and A549 lung cancer cells. Both compounds showed
antiproliferative effects which correlate with their SIRT2 inhibition
potency. SIRT2 is likely to be the target in the cancer cell lines, as
we could show that the degree of acetylation of α-tubulin increased in a
dose-dependent manner. A homology model of SIRT2 based on a SIRT1
crystal structure was built, and docking studies clarified the binding
mode of the chroman-4-one-based inhibitors. The proposed binding mode of
our compounds was similar to other reported SIRT inhibitors in that
they occupy the nicotinamide binding site and prevent NAD+ to
bind in a catalytically active conformation. The docking studies
contributed also to a deeper understanding of the SAR data. However, the
reasons behind the isoform selectivity of the chroman-4-ones are still
unclear but work is ongoing to get an understanding of the selectivity
profile and to verify the binding mode.
Experimental Section
General
All
reactions were carried out using magnetic stirring under ambient
atmosphere if not otherwise noted. Room temperature corresponds to a
temperature interval from 20 to 21 °C. All starting materials and
reagents were obtained from commercial producers and were used without
prior purification. Solvents were generally used as supplied by the
manufacturer. Microwave reactions were carried out using a Biotage
Initiator Sixty with fixed hold time modus in 0.5–2, 2–5 mL, or 10–20 mL
capped microwave vials. All reactions were monitored by thin-layer
chromatography (TLC) on silica plated aluminum sheets (Silica gel 60
F254, E. Merck). Spots were detected by UV light (254 or 365 nm).
Purification by flash column chromatography was performed using an
automatic Biotage SP4 Flash+ instrument. Prefabricated columns of two
different cartridge sizes (surface area 500 m2/g, porosity 60
Å, particle size 40–63 μm) were used. The NMR spectra were measured
with a JEOL JNM-ECP 400 or a Varian 400-MR spectrometer. 1H and 13C
NMR spectra were measured at 400 and 100 MHz, respectively. Chemical
shifts are reported in ppm with the solvent residual peak as internal
standard (CDCl3 δH 7.26, δC 77.16; CD3OD δH 3.31, δC 49.00; acetone-d6 δH 2.05, δC 29.84; DMSO-d6 δH 2.50, δC
39.52). All NMR experiments were measured at ambient temperature.
Melting points were measured with a Mettler FP82 hot stage equipped with
a FP80 temperature controller or Büchi Melting point B-545 and are
uncorrected. Positive ion mass spectra (ESI-MS) were acquired with an
LCQ quadrupole ion trap mass spectrometer (Finnigan LTQ) equipped with
an electrospray ionization source or on a PerkinElmer API 150EX mass
spectrometer. Combustion analyses for CHN were measured on a Thermo
Quest CE Instruments EA 1110 CHNS-O elemental analyzer. High-resolution
mass spectrometry (HRMS) analysis was performed on a Waters LCTp XE mass
spectrometer with an Acquity UPLC BEH C18 (pH 10) or an Acquity UPLC
CSH C18 (pH 3) column eluting with a gradient of 5–95% acetonitrile in
water confirming ≥95% purity. Waters MassLynx 4.1 software was used for
data analysis. Compounds 1, 2, and 20–23 have been synthesized according to procedures earlier reported by our group(32, 40) and by Ankner et al.(41)
General Procedure for the Swern Oxidation to Obtain Aldehydes 4a–b,d–g
DMSO
(3 equiv) was added dropwise to a solution of oxalyl chloride (1.2
equiv) in dry THF (0.1 M) at −78 °C under inert atmosphere, and the
mixture was stirred for 30 min. The appropriate alcohol (1 equiv) in dry
THF (0.5 M) was added dropwise to the reaction mixture, which was
stirred for an additional 30 min at −78 °C. Et3N (5 equiv) was added dropwise, and the mixture was stirred for 15 min before it was allowed to reach room temperature.
Workup Procedure A
Water
and EtOAc were added, and the phases were separated. The aqueous phase
was extracted with EtOAc, and the combined organic phases were washed
with water and brine, dried over MgSO4, and filtered, and the solvent was removed under reduced pressure.
Workup Procedure B
The precipitate was filtered off and rinsed thoroughly with EtOAc. The filtrate was concentrated under reduced pressure.
Aldehydes 4a–g were directly used in the next step without further purification and full characterization.
4-(tert-Butyldimethylsilyloxy)butanal (4a)
The aldehyde was synthesized according to the general procedure from 4-(tert-butyldimethylsilyl)oxy-1-butanol (1.01 g, 4.93 mmol), DMSO (1.0 mL, 14.1 mmol), oxalyl chloride (0.5 mL, 5.73 mmol), and Et3N (3.4 mL, 24.4 mmol). Workup procedure A was used to afford 4a (974 mg). The 1H NMR spectrum of the crude product was in agreement with data reported in the literature.(71)
5-(tert-Butyldimethylsilyloxy)pentanal (4b)
The aldehyde was synthesized according to the general procedure from 5-(tert-butyldimethylsilyl)oxy-1-pentanol 3b (2.07 g, 9.48 mmol), DMSO (2.0 mL, 28.4 mmol), oxalyl chloride (1.0 mL, 11.4 mmol), and Et3N (6.6 mL, 47.4 mmol). Workup procedure A was used to afford 4b (2.00 g). The 1H NMR spectrum of the crude product was in agreement with data reported in the literature.(72)
6-(tert-Butyldimethylsilyloxy)hexanal (4c)
To a suspension of Dess–Martin periodinane (2.06 g, 4.86 mmol) in dry CH2Cl2 (10 mL) at 0 °C was dropwise added a solution of 3c (0.75 g, 3.24 mmol) in dry CH2Cl2
(22 mL). The mixture was allowed to reach room temperature and was
stirred for 1.5 h. The amount of solvent was reduced to half, the
remaining mixture was diluted with Et2O, and an aqueous solution of Na2S2O3/NaHCO3
was added. After 15 min, the phases were separated and the aqueous
phase was extracted with EtOAc. The combined organic phases were washed
with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The 1H NMR spectrum of the crude product was in agreement with data reported in the literature.(73)
2-(2-Methoxyethoxy)acetaldehyde (4d)
The
aldehyde was synthesized according to the general procedure from
2-(2-methoxyethoxy)ethanol (1.52 g, 12.6 mmol), DMSO (2.69 mL, 37.9
mmol), oxalyl chloride (1.32 mL, 15.2 mmol), and Et3N (8.8 mL, 63.2 mmol). Workup procedure B was used to afford 4d (2.83 mg).
3-(Pyridin-2-yl)propanal (4e)
The
aldehyde was synthesized according to the general procedure from
3-(pyridine-2-yl)propan-1-ol (71 mg, 0.52 mmol), DMSO (0.11 mL, 1.55
mmol), oxalyl chloride (0.50 mL, 0.62 mmol), and Et3N (0.36 mL, 2.59 mmol). Workup procedure A was used to afford 4e (70 mg). The 1H NMR spectrum of crude product was in agreement with data reported in the literature.(74)
3-(Pyridin-3-yl)propanal (4f)
The
aldehyde was synthesized according to the general procedure from
3-(pyridine-3-yl)propan-1-ol (222 mg, 1.62 mmol), DMSO (0.34 mL, 4.85
mmol), oxalyl chloride (0.17 mL, 1.94 mmol), and Et3N (1.13 mL, 8.10 mmol). Workup procedure A was used to afford 4f (420 mg). The 1H NMR spectrum of the crude product was in agreement with data reported in the literature.(74)
3-(Pyridin-4-yl)propanal (4g)
The
aldehyde was synthesized according to the general procedure from
3-(pyridine-4-yl)propan-1-ol (136 mg, 0.99 mmol), DMSO (0.21 mL, 2.97
mmol), oxalyl chloride (0.10 mL, 1.19 mmol), and Et3N (0.69 mL, 4.96 mmol). Workup procedure A was used to afford 4g (263 mg). The 1H NMR spectrum of the crude product was in agreement with data reported in the literature.(74)
General Procedure for Synthesis of Chroman-4-ones 6a–i
The
appropriate aldehyde (1.0 equiv) (commercially available or synthesized
from the corresponding alcohol as mentioned above) and DIPA (1.1 equiv)
were added to a 0.4 M solution of the appropriate
2′-hydroxyacetophenone (1.1 equiv) in EtOH. The mixture was heated by
microwave irradiation at 170 °C for 1–2 h (fixed hold time, normal
absorption), and the solvent was removed in vacuo. The residue was
dissolved in EtOAc and washed with 10% NaOH (aq), 1 M HCl (aq), water,
and finally with brine. The organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography gave chroman-4-ones 6d–i. For the synthesis of 6a–c, the corresponding TBDMS-protected chroman-4-ones 5a–c
were dissolved in MeOH (0.1 M), Selectfluor (0.2 equiv) was added, and
the mixture was heated by microwave irradiation to 150 °C for 30 min.
The mixture was concentrated and purified by flash column chromatography
to give chroman-4-ones 6a–c.
8-Bromo-6-chloro-2-(3-hydroxypropyl)chroman-4-one (6a)
The compound was synthesized according to the general procedure from 4a (crude, 974 mg), 3′-bromo-5′-chloro-2′-hydroxyacetophenone (1.28 g, 5.13 mmol), and DIPA (1 mL, 7.1 mmol) to give 5a
(2.00 g). An aliquot (460 mg, 1.06 mmol) was further reacted with
Selectfluor (77 mg, 0.22 mmol). Flash column chromatography was
performed using EtOAc/heptane (2:8 → 6:4) as eluent to afford 6a (280 mg, 78% over three steps) as a white solid; mp 88–89 °C. 1H NMR (CDCl3) δ 7.78 (d, J = 2.5 Hz, 1H), 7.68 (d, J = 2.5 Hz, 1H), 4.64–4.48 (m, 1H), 3.83–3.70 (m, 2H), 2.79–2.66 (m, 2H), 2.10–1.75 (m, 4H). 13C NMR (CDCl3) δ 190.5, 156.6, 138.5, 127.1, 125.9, 122.4, 112.8, 79.0, 62.2, 42.6, 31.3, 28.3. Anal. (C12H12BrClO3) C, H, N.
8-Bromo-6-chloro-2-(4-hydroxybutyl)chroman-4-one (6b)
The compound was synthesized according to the general procedure from 4b
(crude, 2.00 g), 3′-bromo-5′-chloro-2′-hydroxy-acetophenone (2.65 g,
10.6 mmol), and DIPA (1.95 mL, 13.8 mmol). The obtained chroman-4-one 5b
(3.04 g) was directly reacted with Selectfluor (480 mg, 1.35 mmol).
Flash column chromatography was performed using EtOAc/heptane (2:8 →
6:4) as eluent to afford 6b (1.78 g, 56% over three steps) as a white solid; mp 94–96 °C. 1H NMR (CDCl3) δ 7.80 (d, J = 2.6 Hz, 1H), 7.70 (d, J = 2.6 Hz, 1H), 4.57–4.48 (m, 1H), 3.71 (t, J = 5.9 Hz, 2H), 2.79–2.66 (m, 2H), 2.05–1.92 (m, 1H), 1.84–1.50 (m, 5H). 13C NMR (CDCl3) 190.6, 156.7, 138.6, 127.1, 125.9, 122.5, 112.9, 79.0, 62.7, 42.5, 34.5, 32.3, 21.6. Anal. (C13H14BrClO3) C, H, N.
8-Bromo-6-chloro-2-(5-hydroxypentyl)chroman-4-one (6c)
The compound was synthesized according to the general procedure from 4c
(crude, 678 mg), 3′-bromo-5′-chloro-2′-hydroxyacetophenone (954 mg, 3.8
mmol), and DIPA (0.45 mL, 3.24 mmol). The obtained chroman-4-one 5c
(805 mg) was further reacted with Selectfluor (123 mg, 0.35 mmol).
Flash column chromatography was performed using EtOAc/heptane (4:6 →
6:4) as eluent to afford 6c (186 mg, 16% over three steps) as a white solid; mp 110–112 °C. 1H NMR (CDCl3) δ 7.80 (d, J = 2.5 Hz, 1H), 7.70 (d, J = 2.6 Hz, 1H), 4.57–4.47 (m, 1H), 3.68 (t, J = 6.5 Hz, 2H), 2.79–2.65 (m, 2H), 2.03–1.90 (m, 1H), 1.81–1.41 (m, 7H). 13C NMR (CDCl3) δ 190.7, 156.8, 138.5, 127.0, 125.9, 122.5, 112.9, 79.0, 62.9, 42.6, 34.7, 32.7, 25.6, 25.0. Anal. (C14H16BrClO3) C, H, N.
8-Bromo-6-chloro-2-((2-methoxyethoxy)methyl)chroman-4-one (6d)
The compound was synthesized according to the general procedure from 4d
(crude, 50 mg), 3′-bromo-5′-chloro-2′-hydroxyacetophenone (106 mg, 0.42
mmol), and DIPA (86 μL, 0.85 mmol). Flash column chromatography was
performed using EtOAc/heptane (3:7) as eluent to afford 6d (24 mg, 16% over two steps) as an off-white solid; mp 61–63 °C. 1H NMR (CDCl3) δ 7.79 (d, J = 2.6 Hz, 1H), 7.69 (d, J = 2.6 Hz, 1H), 4.74–4.64 (m, 1H), 3.94–3.72 (m, 4H), 3.56 (t, J = 4.6 Hz, 2H), 3.37 (s, 3H), 2.91 (dd, J = 17.1, 12.4 Hz, 1H), 2.76 (dd, J = 17.1, 3.3 Hz, 1H). 13C NMR (CDCl3) δ 190.2, 156.5, 138.5, 127.2, 125.8, 122.4, 112.7, 78.3, 72.4, 72.1, 71.6, 59.2, 39.0. Anal. (C13H14BrClO4) C, H, N.
8-Bromo-6-chloro-2-(2-(pyridine-2-yl)ethyl)chroman-4-one (6e)
The compound was synthesized according to the general procedure from 4e
(crude, 70 mg), 3′-bromo-5′-chloro-2′-hydroxyacetophenone (130 mg, 0.53
mmol), and DIPA (0.1 mL, 0.71 mmol). Flash column chromatography was
performed using EtOAc/heptane (2:8 → 55:45) as eluent to afford 6e (102 mg, 54% over two steps) as a gray–black solid; mp 68–70 °C. 1H NMR (CDCl3) δ 8.54 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 7.79 (d, J = 2.5 Hz, 1H), 7.71 (d, J = 2.6 Hz, 1H), 7.60 (ddd, J = 7.8, 7.5, 1.9 Hz, 1H), 7.24 (ddd, J = 7.8, 1.1, 0.9 Hz, 1H), 7.13 (ddd, J = 7.5, 4.9, 1.1 Hz, 1H), 4.57–4.44 (m, 1H), 3.20–3.01 (m, 2H), 2.81–2.69 (m, 2H), 2.42–2.20 (m, 2H). 13C NMR (CDCl3) δ 190.5, 160.3, 156.6, 149.6, 138.5, 136.7, 127.1, 125.9, 123.4, 122.5, 121.6, 112.9, 78.2, 42.6, 34.2, 33.5. Anal. (C16H13BrClNO2) C, H, N.
8-Bromo-6-chloro-2-(2-(pyridin-3-yl)ethyl)chroman-4-one (6f)
The compound was synthesized according to the general procedure from 4f
(crude, 420 mg), 3′-bromo-5′-chloro-2′-hydroxyacetophenone (445 mg,
1.78 mmol), and DIPA (0.34 mL, 2.41 mmol). Flash column chromatography
was performed using EtOAc/heptane (20:80 → 55:45) as eluent to afford 6f (289 mg, 49% over two steps) as a yellow solid; mp 81–82 °C. 1H NMR (CDCl3) δ 8.54 (d, J = 2.2 Hz, 1H), 8.47 (dd, J = 4.9, 1.6 Hz, 1H), 7.80 (d, J = 2.6 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.58 (dt, J = 7.8, 1.9 Hz, 1H), 7.24 (ddd, J = 7.8, 4.8, 0.8 Hz, 1H), 4.49–4.37 (m, 1H), 3.08–2.86 (m, 2H), 2.80–2.65 (m, 2H), 2.36–2.22 (m, 1H), 2.06–1.93 (m, 1H). 13C NMR (CDCl3) δ 190.1, 156.4, 150.2, 148.1, 138.7, 136.2, 135.9, 127.4, 126.0, 123.6, 122.5, 112.9, 77.3, 42.5, 36.1, 28.4. Anal. (C16H13BrClNO2) C, H, N.
8-Bromo-6-chloro-2-(2-(pyridin-4-yl)ethyl)chroman-4-one (6g)
The compound was synthesized according to the general procedure from an aliquot of 4g
(crude, 134 mg), 3′-bromo-5′-chloro-2′-hydroxyacetophenone (247 mg,
0.99 mmol), and DIPA (0.15 mL, 1.06 mmol). Flash column chromatography
was performed using EtOAc/heptane (3:7 → 100% EtOAc) as eluent to afford
6g (200 mg, 29% over two steps) as an off-white solid; mp 121–123 °C. 1H NMR (CDCl3) δ 8.53 (app d, 2H), 7.81 (d, J = 2.6 Hz, 1H), 7.74 (d, J
= 2.6 Hz, 1H), 7.20 (app d, 2H), 4.49–4.38 (m, 1H), 3.07–2.87 (m, 2H),
2.81–2.69 (m, 2H), 2.36–2.23 (m, 1H), 2.08–1.95 (m, 1H). 13C NMR (CDCl3) δ 190.0, 156.4, 150.1, 149.6, 138.7, 127.4, 126.0, 124.1, 122.5, 112.8, 77.4, 42.5, 35.3, 30.6. Anal. (C16H13BrClNO2) C, H, N.
6-Bromo-2-pentylchroman-4-one (6h)
The
compound was synthesized according to the general procedure from
5′-bromo-2′-hydroxyacetophenone (504 mg, 2.34 mmol), hexanal (0.31 mL,
2.58 mmol), and DIPA (0.37 mL, 2.63 mmol). Flash column chromatography
was performed using toluene/heptane (1:1) as eluent to afford 6h (433 mg, 62%) as a white solid; mp 42–44 °C. 1H NMR (CDCl3) δ 7.97 (d, J = 2.5 Hz, 1H), 7.53 (dd, J = 8.8, 2.5 Hz, 1H), 6.88 (d, J
= 8.8 Hz, 1H), 4.47–4.37 (m, 1H), 2.74–2.60 (m, 2H), 1.93–1.81 (m, 1H),
1.75–1.64 (m, 1H), 1.60–1.25 (m, 6H), 0.95–0.87 (m, 3H). 13C NMR (CDCl3) δ 191.5, 160.7, 138.7, 129.5, 122.4, 120.2, 113.9, 78.3, 42.7, 34.9, 31.7, 24.7, 22.7, 14.1. Anal. (C14H17BrO2) C, H, N.
6-Bromo-8-chloro-2-pentylchroman-4-one (6i)
The
compound was synthesized according to the general procedure from
5′-bromo-3′-chloro-2′-hydroxyacetophenone (498 mg, 2.00 mmol), hexanal
(0.24 mL, 2.00 mmol), and DIPA (0.34 mL, 2.39 mmol). Flash column
chromatography was performed using EtOAc/heptane (5:95) as eluent to
afford 6i (364 mg, 55%) as a white solid; mp 75–77 °C. 1H NMR (CDCl3) δ 7.90 (d, J = 2.4 Hz, 1H), 7.67 (d, J
= 2.4 Hz, 1H), 4.56–4.46 (m, 1H), 2.79–2.65 (m, 2H), 2.01–1.88 (m, 1H),
1.79–1.67 (m, 1H), 1.67–1.29 (m, 6H), 0.95–0.85 (m, 3H).13C NMR (CDCl3) δ 190.6, 156.4, 138.2, 128.2, 124.5, 123.2, 113.2, 79.2, 42.6, 34.7, 31.6, 24.7, 22.6, 14.1. Anal. Calcd for C14H16BrClO2: C, 50.70; H, 4.86. Found: C, 51.42; H, 4.86.
1-(3-Bromo-5-chloro-2-hydroxyphenyl)-2-(tetrahydrofuran-2-yl)ethanone (7a)
A solution of TBAF in THF (1 M, 0.18 mL, 0.18 mmol) was added to a stirred solution of 5a
(53 mg, 0.12 mmol) in dry THF (11 mL), the reaction mixture was stirred
at room temperature overnight. The mixture was concentrated under
reduced pressure, and the crude product was purified by flash column
chromatography using EtOAc/heptane (1:9) as eluent to afford 7a (30 mg, 76%) as a pale-yellow oil. 1H NMR (CDCl3) δ 12.85 (s, 1H), 7.72 (s, 2H), 4.43–4.34 (m, 1H), 3.93–3.85 (m, 1H), 3.80–3.72 (m, 1H), 3.32 (dd, J = 16.1, 7.0 Hz, 1H), 3.05 (dd, J = 16.1, 5.4 Hz, 1H), 2.25–2.12 (m, 1H), 2.00–1.88 (m, 2H), 1.65–1.52 (m, 1H). 13C NMR (CDCl3) δ 203.8, 157.9, 139.0, 129.1, 124.0, 120.6, 113.1, 75.0, 68.2, 44.6, 31.8, 25.7.
1-(3-Bromo-5-chloro-2-hydroxyphenyl)-2-(tetrahydropyran-2-yl)ethanone (7b)
A solution of TBAF in THF (1 M, 1.3 mL, 1.32 mmol) was added to a stirred solution of 5b
(197 mg, 0.44 mmol) in dry THF (2 mL), and the reaction mixture was
stirred at room temperature for 17 h. The mixture was concentrated under
reduced pressure, and the crude product was purified by flash
chromatography using EtOAc/heptane (8:92) as eluent to afford 7b (106 mg, 72%) as a yellow oil. 1H NMR (CDCl3) δ 12.90 (s, 1H), 7.74 (d, J = 2.5 Hz, 1H), 7.72 (d, J = 2.5 Hz, 1H), 3.97–3.83 (m, 2H), 3.50–3.37 (m, 1H), 3.26 (dd, J = 15.7, 7.7 Hz, 1H), 2.88 (dd, J = 15.7, 4.6 Hz, 1H), 1.94–1.81 (m, 1H), 1.75–1.29 (m, 5H). 13C NMR (CDCl3) δ 203.9, 157.9, 139.0, 129.3, 123.9, 120.9, 113.0, 74.2, 68.8, 45.2, 32.0, 25.8, 23.4.
Methyl 4-Oxobutanoate (8a)
Et3N
(0.24 mL, 1.73 mmol) was added to a solution of γ-butyrolactone (0.40
mL, 5.20 mmol) in MeOH (5 mL). The mixture was stirred overnight at room
temperature. Toluene was added, and the solvent was removed under
reduced pressure to give the crude methyl 4-hydroxybutanoate as a
colorless liquid. DMSO (0.74 mL, 10.4 mmol) was added dropwise to a
solution of oxalyl chloride (0.36 mL, 4.16 mmol) in dry THF (21 mL) at
−78 °C under inert atmosphere, and the mixture stirred for 30 min.
Methyl 4-hydroxybutanoate (410 mg, 3.47 mmol) in dry THF (7 mL) was
added dropwise to the reaction mixture which was stirred for an
additional 30 min at −78 °C. Et3N (2.42 mL, 17.3 mmol) was
added dropwise, and the mixture was stirred for 15 min and was then
allowed to reach room temperature. Water and EtOAc were added, and the
phases were separated. The aqueous phase was extracted with EtOAc, and
the combined organic phases were washed with water and brine, dried over
MgSO4, and filtered, and the solvent was removed under reduced pressure to give 8a
(293 mg, 43% over two steps). The crude product was sufficiently pure
to be used in the next step without further purification. The 1H NMR spectrum of crude product was in agreement with data reported in the literature.(75)
Methyl 5-Hydroxypentanoate (8b)
Et3N
(2.0 mL, 14 mmol) was added to a solution of δ-valerolactone (4.2 g, 42
mmol) in MeOH (40 mL). The mixture was stirred for 18 h at room
temperature. Toluene was added, and the solvent was removed under
reduced pressure. A part of the crude alcohol (2.7 g, 20 mmol) and Et3N (8.2 mL, 59 mmol) were dissolved in dry DMSO (40 mL) under inert atmosphere. A solution of SO3·pyridine
(9.3 g, 59 mmol) in DMSO (30 mL) was added dropwise, and the mixture
was stirred for 14 h at room temperature. The mixture was poured on
brine (400 mL) and ice (100 mL), and the product was extracted with CH2Cl2 and EtOAc. The combined organic phases were dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography using EtOAc/hexane (2:8) gave 8b (1.68 g, 63% over two steps). The 1H NMR spectrum of crude product was in agreement with data reported in the literature.(76)
Methyl 3-(8-Bromo-6-chloro-4-oxochroman-2-yl)propanoate (9a)
3′-Bromo-5′-chloro-2′-hydroxyacetophenone (591 mg, 2.37 mmol) was dissolved in EtOH (10 mL), DIPA (0.36 mL, 2.58 mmol) and 8a
(250 mg, 2.15 mmol) were added to a microwave vial, and the mix was
heated in the microwave to 170 °C for 1 h. The solvent was removed, and
the residue was redissolved in EtOAc. The organic phase was washed with
0.1 M HCl (aq), 1% and 10% NaOH (aq), water, and brine. The organic
phase was dried over MgSO4 and filtered, and the solvent was
removed under reduced pressure. Purification by flash chromatography
using EtOAc:pentane (1:4) gave 9a (300 mg, 40%) as an off-white solid; mp 102–104 °C. 1H NMR (CDCl3) δ 7.81 (d, J = 2.3 Hz, 1H), 7.71 (d, J = 2.3 Hz, 1H), 4.65–4.50 (m, 1H), 3.72 (s, 3H), 2.89–2.53 (m, 4H), 2.30–2.01 (m, 2H). 13C NMR (CDCl3) δ 190.1, 173.2, 156.4, 138.6, 127.3, 126.0, 122.5, 112.9, 77.9, 52.0, 42.5, 29.9, 29.5. Anal. (C13H12BrClO4) C, H, N.
Methyl 4-(8-Bromo-6-chloro-4-oxochroman-2-yl)butanoate (9b)
3′-Bromo-5′-chloro-2′-hydroxyacetophenone (242 mg, 0.97 mmol), 8b
(139 mg, 1.07 mmol), and piperidine (0.01 mL, 0.97 mmol) were added to a
microwave vial followed by EtOH (2 mL). The mixture was heated by
microwave irradiation to 170 °C for 30 min. The solvent was removed
under reduced pressure. Purification by flash chromatography using
EtOAc/hexane (12:88 and 2:8) gave 9b (225 mg, 64%) as a yellow solid; mp 64–66 °C. 1H NMR (CD3OD) δ 7.82 (d, J = 2.5 Hz, 1H), 7.75 (d, J = 2.6 Hz, 1H), 4.65–4.55 (m, 1H), 3.67 (s, 3H), 2.85–2.69 (m, 2H), 2.47 (t, J = 7.0 Hz, 2H), 2.07–1.73 (m, 4H). 13C NMR (CD3OD) δ 192.1, 175.5, 158.1, 139.2, 127.7, 126.4, 123.8, 113.8, 80.3, 52.0, 43.1, 34.9, 34.3, 21.8. Anal. (C14H14BrClO4) C, H, N.
3-(8-Bromo-6-chloro-4-oxo-chroman-2-yl)propanoic acid (10a)
To a solution of 6a (378 mg, 1.18 mmol) in dry CH2Cl2
(15 mL) Dess–Martin periodinane (785 mg, 1.80 mmol) was added. The
mixture was stirred for 1 h at room temperature. The reaction was
quenched by the addition of 10% Na2S2O3/NaHCO3 (aq). After 5 min, the mix was diluted with CH2Cl2 and H2O and the aqueous phase was extracted with CH2Cl2. The combined organic phases were washed with brine, dried over MgSO4,
filtered, and concentrated under reduced pressure. The crude aldehyde
(387 mg) was dissolved in THF (30 mL) and cooled to 0 °C. Amylene (1.25
mL, 11.8 mmol) was added, and NaClO2 (321 mg, 3.55 mmol) and NaH2PO4·2H2O (371 mg, 2.37 mmol) dissolved in H2O
(14 mL) were added dropwise. The ice bath was removed, and the mixture
was stirred for 1 h. The reaction was quenched by the addition of a
mixture of 1 M HCl and brine (1:1) and EtOAc. After 5 min of stirring,
the phases were separated and the aqueous phase was extracted with
EtOAc. The combined organic phases were washed with 1 M HCl/brine mix.
The organic phase was extracted with 0.1 M NaOH (aq). The basic aqueous
phase was acidified with 1 M HCl, and the acidic aqueous phase was
extracted with EtOAc. The combined organic phases were finally washed
with brine, dried over MgSO4, filtered, and concentrated
under reduced pressure. Flash column chromatography was performed using
EtOAc/heptane (3:7) with 1% AcOH as eluent to afford 10a (290 mg, 73% over two steps) as a white solid; mp 177–179 °C. 1H NMR (CD3OD) δ 7.84 (d, J = 2.6 Hz, 1H), 7.76 (d, J = 2.6 Hz, 1H), 4.70–4.58 (m, 1H), 2.90–2.54 (m, 4H), 2.21–2.03 (m, 2H). 13C NMR (CD3OD) δ 191.8, 176.4, 157.9, 139.2, 127.8, 126.4, 123.7, 113.9, 79.6, 43.0, 30.9, 30.4. Anal. (C12H10BrClO4) C, H, N.
4-(8-Bromo-6-chloro-4-oxo-chroman-2-yl)butanoic Acid (10b)
To a solution of 6b (935 mg, 2.80 mmol) in dry CH2Cl2
(40 mL) Dess–Martin periodinane (1.82 g, 4.16 mmol) was added. The
mixture was stirred for 45 min at room temperature. The reaction was
quenched by the addition of 10% Na2S2O3/NaHCO3 (aq). After 5 min, the mix was diluted with CH2Cl2 and H2O and the aqueous phase was extracted with CH2Cl2. The combined organic phases were washed with brine, dried over MgSO4,
filtered, and concentrated under reduced pressure. The crude aldehyde
(1.14 g) was dissolved in THF (70 mL) and cooled to 0 °C. Amylene (1.96
g, 28.0 mmol) was added, and NaClO2 (762 mg, 8.42 mmol) and NaH2PO4·2H2O (875 mg, 5.61 mmol) dissolved in H2O
(33 mL) were added dropwise. The ice bath was removed, and the mixture
was stirred for 2.5 h. The reaction was quenched by the addition of a
mixture of 1 M HCl and brine (1:1) and EtOAc. After 5 min of stirring,
the phases were separated and the aqueous phase was extracted with
EtOAc. The combined organic phases were washed with brine/1 M HCl mix.
The organic phase was extracted with 0.1 M NaOH. The basic aqueous phase
was acidified with 1 M HCl (aq), and the acidic aqueous phase was
extracted with EtOAc. The combined organic phases were finally washed
with brine, dried over MgSO4, filtered, and concentrated
under reduced pressure. Flash chromatography was performed using
EtOAc/heptane (3:7) with 1% AcOH to afford 10b (716 mg, 74% over two steps) as an off-white solid; mp 123–124 °C. 1H NMR (CD3OD) δ 7.83 (d, J = 2.6 Hz, 1H), 7.75 (d, J = 2.5 Hz, 1H), 4.66–4.56 (m, 1H), 2.85–2.70 (m, 2H), 2.43 (t, J = 7.1 Hz, 2H), 2.05–1.76 (m, 4H). 13C NMR (CD3OD) δ. 192.1, 177.1, 158.2, 139.2, 127.7, 126.4, 123.8, 113.9, 80.3, 43.1, 35.0, 34.4, 21.8. Anal. (C13H12BrClO4) C, H, N.
General Procedure for the Synthesis of Amides 11a–e
A 0.14 M solution of the appropriate carboxylic acid (1 equiv) in dry CH2Cl2 containing 5–10 vol % DMF was cooled to 0 °C under inert atmosphere. N,N′-Carbonyldiimidazole
(1.5 equiv) was added, and the mixture was stirred for 30 min. The
appropriate amine (3 equiv) was added, and the mixture was stirred at
room temperature for 2–14 h. The mixture was diluted with CH2Cl2 and washed with 1 M HCl (aq) and brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography gave the amides 11a–e.
3-(8-Bromo-6-chloro-4-oxochroman-2-yl)-N-methylpropanamide (11a)
The title compound was synthesized according to the general procedure from 10a (99 mg, 0.30 mmol), methylamine hydrochloride (62 mg, 0.91 mmol), and N,N′-carbonyldiimidazole (73 mg, 1.52 mmol). Flash chromatography was performed using MeOH/CH2Cl2 (3:97) to afford 11a (70 mg, 68%) as a white solid; mp 178–180 °C. 1H NMR (CDCl3) δ 7.81 (d, J = 2.4 Hz, 1H), 7.70 (d, J = 2.5 Hz, 1H), 5.63 (br s, 1H), 4.61–4.51 (m, 1H), 2.86–2.66 (m, 5H), 2.51 (app t, 2H), 2.26–2.07 (m, 2H). 13C NMR (CDCl3) δ 190.1, 172.2, 156.3, 138.5, 127.3, 126.1, 122.6, 112.6, 78.0, 42.5, 31.6, 30.4, 26.6. Anal. (C13H13BrClNO3) C, H, N.
4-(8-Bromo-6-chloro-4-oxochroman-2-yl)-N-methylbutanamide (11b)
The title compound was synthesized according to the general procedure from 10b (100 mg, 0.28 mmol), methyl amine hydrochloride (60 mg, 0.89 mmol), and N,N′-carbonyldiimidazole (72 mg, 0.44 mmol). Flash chromatography was performed using MeOH/CH2Cl2 (3:97) to afford 2b (96 mg, 93%) as a white solid; mp 136–139 °C. 1H NMR (CDCl3) δ 7.78 (d, J = 2.6 Hz, 1H), 7.69 (d, J = 2.6 Hz, 1H), 5.62 (s, 1H), 4.59–4.46 (m, 1H), 2.81 (d, J = 4.6 Hz, 3H), 2.76–2.66 (m, 2H), 2.36–2.24 (m, 2H), 2.03–1.75 (m, 4H). 13C NMR (CDCl3) δ 190.4, 173.0, 156.6, 138.5, 127.1, 125.9, 122.5, 112.7, 79.0, 42.5, 35.8, 34.1, 26.5, 21.4. Anal. Calcd for C14H15BrClNO3 C, 46.63; H, 4.19; N, 3.88. Found: C, 47.28; H, 4.23; N, 3.78.
4-(8-Bromo-6-chloro-4-oxochroman-2-yl)-N-isopropylbutanamide (11c)
The title compound was synthesized according to the general procedure from 10b (100 mg, 0.28 mmol), iso-propylamine (76 μL, 0.89 mmol), and N,N′-carbonyldiimidazole (72 mg, 0.44 mmol). Flash chromatography was performed using MeOH/CH2Cl2 (3:97) to afford 11c (97 mg, 84%) as an off-white solid; mp 151–154 °C. 1H NMR (CDCl3) δ 7.81 (d, J = 2.6 Hz, 1H), 7.70 (d, J
= 2.5 Hz, 1H), 5.29 (s, 1H), 4.63–4.44 (m, 1H), 4.17–4.00 (m, 1H),
2.79–2.66 (m, 2H), 2.32–2.22 (m, 2H), 2.05–1.75 (m, 4H), 1.15 (d, J = 6.5 Hz, 6H). 13C NMR (CDCl3) δ 190.5, 171.4, 156.6, 138.5, 127.2, 126.0, 122.5, 112.8, 79.1, 42.5, 41.5, 36.1, 34.0, 23.02, 23.00, 21.5. Anal. (C16H19BrClNO3) C, H, N.
N-Benzyl-4-(8-bromo-6-chloro-4-oxochroman-2-yl)butanamide (11d)
The title compound was synthesized according to the general procedure from 10b (85 mg, 0.24 mmol), benzyl amine (80 μL, 0.73 mmol), and N,N′-carbonyldiimidazole
(60 mg, 0.37 mmol). Flash chromatography was performed using
EtOAc/heptane (1:1 → 7:3 stepwise) and EtOAc/heptane (6:4 → 8:2) to
afford 11d (81 mg, 76%) as a pale-yellow solid; mp 128–131 °C. 1H NMR (CDCl3) δ 7.79 (d, J = 2.6 Hz, 1H), 7.69 (d, J = 2.6 Hz, 1H), 7.37–7.23 (m, 5H), 5.85 (s, 1H), 4.57–4.47 (m, 1H), 4.45 (d, J = 5.6 Hz, 2H), 2.76–2.64 (m, 2H), 2.42–2.30 (m, 2H), 2.10–1.75 (m, 4H). 13C NMR (CDCl3)
δ 190.4, 172.2, 156.6, 138.5, 138.3, 128.9, 128.0, 127.7, 127.2, 126.0,
122.5, 112.8, 79.0, 43.8, 42.5, 35.9, 34.0, 21.5. HRMS [M + H]+ calcd for C20H19BrClNO3, 436.0315; found, 436.0315.
4-(8-Bromo-6-chloro-4-oxochroman-2-yl)-N,N-dimethylbutanamide (11e)
The title compound was synthesized according to the general procedure from 10b (100 mg, 0.28 mmol), dimethylamine hydrochloride (72 mg, 0.89 mmol), and N,N′-carbonyldiimidazole (71 mg, 0.44 mmol). Flash chromatography was performed using MeOH/CH2Cl2 (2:98) to afford 11e (82 mg, 76%) as a colorless oil. 1H NMR (CDCl3) δ 7.77 (d, J = 2.6 Hz, 1H), 7.68 (d, J = 2.7 Hz, 1H), 4.59–4.47 (m, 1H), 3.01 (s, 3H), 2.94 (s, 3H), 2.78–2.65 (m, 2H), 2.50–2.35 (m, 2H), 2.04–1.78 (m, 4H). 13C NMR (CDCl3) δ 190.5, 172.3, 156.7, 138.4, 127.0, 125.9, 122.4, 112.8, 79.1, 42.4, 37.3, 35.5, 34.3, 32.7, 20.7. Anal. (C15H17BrClNO3) C, H, N.
8-Bromo-6-chloro-2-(2-(3-methyl-1,2,4-oxadiazol-5-yl)ethyl)chroman-4-one (12a)
To a solution of 10a (100 mg, 0.30 mmol) in MeCN (3 mL) and DMF (0.6 mL) were added N,N′-carbonyldiimidazole
(73 mg, 0.45 mmol) and acetamide oxime (34 mg, 0.45 mmol). The mixture
was heated to 85 °C for 19 h. EtOAc and water were added, and the
aqueous phase was extracted with EtOAc. The combined organic phases were
washed with 1% NaOH (aq) and brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography was performed using MeOH/CH2Cl2 (2:98) to afford 12a (76 mg, 68%) as a yellow oil. 1H NMR (CDCl3) δ 7.79 (d, J = 2.6 Hz, 1H), 7.70 (d, J = 2.5 Hz, 1H), 4.71–4.57 (m, 1H), 3.31–3.11 (m, 2H), 2.82–2.71 (m, 2H), 2.48–2.35 (m, 1H), 2.37 (s, 3H), 2.34–2.23 (m, 1H). 13C NMR (CDCl3) δ 189.7, 178.3, 167.3, 156.2, 138.6, 127.5, 125.9, 122.4, 112.9, 77.4, 42.4, 31.3, 22.4, 11.7. Anal. (C14H12BrClN2O3) C, H, N.
8-Bromo-6-chloro-2-(3-(3-methyl-1,2,4-oxadiazol-5-yl)propyl)chroman-4-one (12b)
To a solution of 10b (154 mg, 0.44 mmol) in MeCN (4.5 mL) and DMF (0.4 mL) were added N,N′-carbonyldiimidazole
(108 mg, 0.66 mmol) and acetamide oxime (50 mg, 0.66 mmol). The mixture
was heated to 85 °C for 14 h. EtOAc and water were added, and the
aqueous phase was extracted with EtOAc. The combined organic phases were
washed with 1% NaOH (aq) and brine, dried over MgSO4, filtered, and concentrated under recued pressure. Flash chromatography was performed using MeOH/CH2Cl2 (3:97) to afford 12b (86 mg, 50%) as a yellow oil. 1H NMR (CDCl3) δ 7.77 (d, J = 2.7 Hz, 1H), 7.68 (d, J = 2.6 Hz, 1H), 4.60–4.48 (m, 1H), 3.08–2.92 (m, 2H), 2.78–2.65 (m, 2H), 2.36 (s, 3H), 2.28–1.94 (m, 3H), 1.93–1.79 (m, 1H). 13C NMR (CDCl3) δ 190.1, 178.9, 167.2, 156.5, 138.5, 127.2, 125.9, 122.4, 112.8, 78.5, 42.4, 33.8, 26.1, 22.3, 11.7. Anal. (C15H14BrClN2O3) C, H, N.
3-Morpholinopropanal (13)
Morpholine (1.63 mL, 18.67 mmol) and acrolein (1.50 mL, 22.42 mmol) were added to a suspension of MgSO4 in MeCN (50 mL), and the reaction mixture was stirred overnight. MgSO4
was filtered off, and the filtrate was concentrated under reduced
pressure. The crude product was coevaporated with MeCN to afford 13 (2.45 g, 93%) as a yellow viscous oil which was used without further purification. 1H NMR (CDCl3) δ 9.77 (t, J = 2.0 Hz, 1H), 3.78–3.53 (m, 6H), 2.76–2.70 (m, 1H), 2.60–2.58 (m, 1H), 2.47–2.41 (m, 4H). 13C NMR (CDCl3) δ 201.6, 67.0, 53.6, 51.8, 41.1.
3-(tert-Butyldimethylsilyloxy)propanal (15)
DMSO (0.60 mL, 8.46 mmol) was added dropwise to a solution of oxalyl chloride (0.37 mL, 4.24 mmol) in dry THF (30 mL) under N2 atmosphere at −78 °C. The reaction mixture was stirred for 40 min, followed by the dropwise addition of 3-(tert-butyldimethylsilyl)oxy-1-propanol
(700 mg, 3.68 mmol) in dry THF (12 mL). The mixture was stirred for
additional 45 min at −78 °C. Et3N (3 mL, 21.52 mmol) was
added dropwise, and the mixture was stirred for 15 min at −78 °C and was
then allowed to warm to room temperature. Water and CH2Cl2 were added, and the phases were separated. The aqueous phase was extracted with CH2Cl2, and the combined organic phases were washed with water and brine, dried over MgSO4, filtered, and concentrated under reduced pressure. 15 (670 mg 97%) was afforded as a colorless oil which was used without further purification. 1H NMR (CDCl3) δ 9.80 (t, J = 2.1 Hz, 1H), 3.99 (t, J = 6.0 Hz, 2H), 2.59 (td, J = 6.0, 2.1 Hz, 2H), 0.88 (s, 9H), 0.06 (s, 6H). 13C NMR (CDCl3) δ 202.1, 57.6, 46.7, 26.0, 18.4, −5.3.
3-(8-Bromo-6-chloro-4-oxochroman-2-yl)propyl methanesulfonate (16)
Mesyl chloride (52 μL, 0.67 mmol) was added to a solution of 6a (150 mg, 0.47 mmol) and Et3N (0.1 mL, 0.72 mmol) in dry CH2Cl2
(4 mL) at 0 °C under inert atmosphere. The mixture was stirred for 2 h
and was then washed with water and brine, dried over MgSO4, filtered, and concentrated under reduced pressure to afford 16 (180 mg, 96%) as a yellow solid. The product was used in the next step without further purification. 1H NMR (CDCl3) δ 7.82 (d, J = 2.5 Hz, 1H), 7.72 (d, J = 2.6 Hz, 1H), 4.60–4.50 (m, 1H), 4.46–4.31 (m, 2H), 3.04 (s, 3H), 2.80–2.69 (m, 2H), 2.26–1.84 (m, 4H). 13C NMR (CDCl3) δ 190.0, 156.4, 138.6, 127.4, 126.0, 122.5, 112.8, 78.4, 69.3, 42.6, 46.1, 37.7, 31.0, 25.4.
8-Bromo-6-chloro-2-(3-morpholinopropyl)chroman-4-one (17a)
Morpholine (50 μL, 0.57 mmol) was added to a solution of 16
(96 mg, 0.24 mmol) in dry THF (2.5 mL). The mixture was heated by
microwave irradiation to 100 °C for 40 min and 150 °C for 25 min. The
mixture was concentrated under reduced pressure, and flash column
chromatography was performed using MeOH/CH2Cl2 (5:95) followed by an acid–base extraction with 1 M HCl and NaOH (aq) to afford 17a (48 mg, 51%) as a yellow oil. 1H NMR (CDCl3) δ 7.78 (d, J = 2.6 Hz, 1H), 7.68 (d, J = 2.6 Hz, 1H), 4.61–4.49 (m, 1H), 3.72–3.66 (m, 4H), 2.79–2.64 (m, 2H), 2.49–2.35 (m, 6H), 2.05–1.62 (m, 4H). 13C NMR (CDCl3) δ 190.5, 156.6, 138.5, 127.0, 125.9, 122.5, 112.8, 77.0, 67.1, 58.4, 53.8, 42.6, 32.6, 22.0. Anal. (C16H19BrClNO3) C, H, N.
8-Bromo-6-chloro-2-(3-(piperidin-1-yl)propyl)chroman-4-one (17b)
Piperidine (60 μL, 0.61 mmol) was added to a solution of 16
(101 mg, 0.25 mmol) in dry THF (2.5 mL). The mixture was heated by
microwave irradiation at 120 °C for 1 h and concentrated under reduced
pressure. Flash column chromatography was performed using MeOH/CH2Cl2 (5:95), followed by an acid–base extraction with 1 M HCl and NaOH (aq) to afford 17b (38 mg, 39%) as a yellow oil. 1H NMR (CDCl3) δ 7.77 (d, J = 2.6 Hz, 1H), 7.68 (d, J = 2.6 Hz, 1H), 4.59–4.48 (m, 1H), 2.80–2.63 (m, 2H), 2.48–2.30 (m, 6H), 2.02–1.32 (m, 10H). 13C NMR (CDCl3) δ 190.6, 156.7, 138.5, 127.0, 125.9, 122.5, 112.8, 79.0, 58.8, 54.6, 42.5, 32.9, 25.9, 24.5, 22.3. Anal. (C17H21BrClNO2) C, H, N.
3-(2-Oxo-1,2-dihydroquinolin-6-yl)propanal (19a)
6-Bromoquinolin-2(1H)-one (303.2 mg, 1.35 mmol), Pd(OAc)2 (31 mg, 0.14 mmol), KCl (102 mg, 1.36 mmol), K2CO3
(282 mg, 2.04 mmol), and TBAA (820 mg, 2.71 mmol) were dissolved in dry
DMF (6 mL) under inert atmosphere. Acrolein diethyl acetal (0.62 mL,
4.06 mmol) was added, and the mixture was heated at 90 °C overnight. The
reaction was divided in three runs. The mixtures were diluted with
EtOAc, filtered through Celite, rinsed with EtOAc, and concentrated
under reduced pressure and coevaporated with toluene to afford 18a as brown oil (1157 mg), which was used in the next step without further purification. 18a was dissolved in MeOH (14 mL), and 10% Pd/C (10 wt %, 111 mg) was added. The mixture was stirred under H2
atmosphere (balloon) for 3 h at room temperature, filtered through
Celite, and the filtrate was concentrated under reduced pressure,
resulting in a brown oil (960 mg). The crude oil was dissolved acetone
(13 mL), whereafter water (0.6 mL) and HCl (0.5 mL, conc) were added.
The mixture was heated to reflux for 4 h. The solvent was removed, and
EtOAc and water were added to the residue. The phases were separated,
and the aqueous phase was extracted with EtOAc. The combined organic
phases were washed with saturated NaHCO3 (aq), water and brine, dried over Na2SO4,
filtered, and concentrated under reduced pressure. The crude product
was purified by flash column chromatography using MeOH/EtOAc (3:97) as
eluent, affording the 19a as a pale-yellow solid (97 mg, 34% over three steps); mp 139–141 °C. 1H NMR (acetone-d6) δ 11.47 (br s, 1H), 9.80 (t, J = 1.3 Hz, 1H), 7.85 (d, J = 9.5 Hz, 1H), 7.52 (d, J = 2.1 Hz, 1H), 7.44 (dd, J = 8.4, 2.0 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 6.55 (d, J = 9.5 Hz, 1H), 2.99 (app t, 2H), 2.86–2.79 (m, 2H). 13C NMR (acetone-d6) δ 202.0, 163.5, 141.1, 138.4, 135.8, 131.9, 128.0, 122.8, 120.5, 116.3, 45.8, 28.0.
3-(Quinolin-6-yl)propanal (19b)
6-Bromoquinoline (106 mg, 0.51 mmol) was dissolved in dry DMF (2 mL) under inert atmosphere, and Pd(OAc)2 (12 mg, 0.054 mmol), KCl (38 mg, 0.51 mmol), K2CO3
(108 mg, 0.78 mmol), and TBAA (311 mg, 1.02 mmol) were added. Acrolein
diethyl acetal (0.23 mL, 1.53 mmol) was added, and the mixture was
heated at 90 °C for 24 h. The reaction was divided in three runs in the
microwave. The mixtures were diluted with EtOAc, filtered through
Celite, rinsed with EtOAc, and concentrated under reduced pressure and
coevaporated with toluene to afford 18b as brown oil (471 mg), which was used in the next step without further purification. 18b
was dissolved EtOH (10 mL), and 1,4-cyclohexadiene (0.48 mL, 10 mmol)
and 10% Pd/C (11 mg, 0.01 mmol) were added. The mixture was heated to
reflux for 1.5 h. Two subsequent additions of 1,4-cyclohexadiene (0.23
mL, 2.5 mmol; 0.48 mL, 5.1 mmol) and 10% Pd/C (11 mg, 0.01 mmol and 8
mg, 0.007 mmol) were made after 1.5 and 3 h of heating. The mixture was
heated to reflux for 4.5 h in total. The mixture was diluted with EtOAc,
filtered through Celite, rinsed with EtOAc, and concentrated under
reduced pressure, affording the crude product as a yellow oil (374 mg).
The crude oil was dissolved acetone (13 mL), whereafter water (2 mL) and
HCl (1 mL, conc) were added. The mixture was heated to reflux for 2 h.
The solvent was removed, and EtOAc and saturated Na2CO3
(aq) were added to the residue. The phases were separated, and the
aqueous phase was extracted with EtOAc. The combined organic phases were
washed with saturated NaHCO3 (aq), water, and brine, dried over MgSO4,
filtered, and concentrated under reduced pressure. The crude product
was purified by flash column chromatography using EtOAc as eluent to
afford 19b as a transparent oil (53 mg, 56% over three steps). 1H NMR (CDCl3) δ 9.82 (t, J = 1.3 Hz, 1H), 8.84 (dd, J = 4.2, 1.8 Hz, 1H), 8.05 (ddd, J = 8.4, 1.9, 0.8 Hz, 1H), 8.01 (d, J = 8.6 Hz, 1H), 7.57 (d, J = 1.4 Hz, 1H), 7.53 (dd, J = 8.6, 2.0 Hz, 1H), 7.34 (dd, J = 8.3, 4.2 Hz, 1H), 3.11 (t, J = 7.5 Hz, 2H), 2.85 (tdd, J = 7.6, 1.3, 0.5 Hz, 2H). 13C NMR (CDCl3) δ 201.1, 150.1, 147.3, 138.8, 135.6, 130.6, 129.8, 128.4, 126.4, 121.3, 45.0, 28.0.
6-(2-(8-Bromo-6-chloro-4-oxochroman-2-yl)ethyl)quinolin-2(1H)-one (20a)
3′-Bromo-5′-chloro-2′-hydroxyacetophenone (58 mg, 0.23 mmol) was dissolved in EtOH (1 mL), DIPA (32 μmL, 0.23 mmol) and 19a
(42 mg, 0.21 mmol) were added to a microwave vial, and the mixture was
heated in the microwave to 160 °C for 1.5 h. The formed brown
precipitate was filtered off, rinsed with EtOAc, and triturated with CH2Cl2, resulting in a pale-yellow solid (44 mg, 49%); mp 229–231 °C. 1H NMR (DMSO-d6) δ 11.67 (br s, 1H), 8.04 (d, J = 2.6 Hz, 1H), 7.83 (d, J = 9.5 Hz, 1H), 7.67 (d, J = 2.6 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.41 (dd, J = 8.4, 2.0 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 6.46 (d, J = 9.5 Hz, 1H), 4.69–4.56 (m, 1H), 3.03–2.72 (m, 4H), 2.25–1.94 (m, 2H). 13C NMR (DMSO-d6)
δ 190.2, 161.8, 156.2, 140.0, 137.7, 137.3, 134.2, 130.9, 127.0, 125.6,
125.0, 122.5, 121.9, 119.1, 115.2, 112.6, 77.8, 41.4, 35.4, 29.8. HRMS
(ESI-LC/MS) [M + H]+: calcd for C20H15BrClNO3, 432.0002; found, 432.0025.
8-Bromo-6-chloro-2-(2-(quinolin-6-yl)ethyl)chroman-4-one (20b)
3′-Bromo-5′-chloro-2′-hydroxyacetophenone (77.1 mg, 0.31 mmol) was dissolved in EtOH (1 mL), DIPA (44 μL, 0.31 mmol) and 19b
(52 mg, 0.28 mmol) were added to a microwave vial, and the mixture was
heated in the microwave reactor to 170 °C for 2 h. The solvent was
removed, and the residue was redissolved in EtOAc. The organic phase was
washed with 10% NaOH (aq), water, and brine, dried over MgSO4,
filtered, and concentrated under reduced pressure. The crude product
was purified by flash column chromatography using EtOAc/pentane (1:1) as
eluent, affording 20b as a pale-yellow solid (18 mg, 15%); mp 187–190 °C. 1H NMR (CDCl3) δ 8.88 (d, J = 4.2 Hz, 1H), 8.10 (dd, J = 8.3, 1.2 Hz, 1H), 8.06 (d, J = 8.7 Hz, 1H), 7.80 (d, J = 2.6 Hz, 1H), 7.74 (d, J = 2.6 Hz, 1H), 7.69 (d, J = 1.9 Hz, 1H), 7.63 (dd, J = 8.6, 2.0 Hz, 1H), 7.39 (dd, J = 8.3, 4.2 Hz, 1H), 4.55–4.41 (m, 1H), 3.30–3.01 (m, 2H), 2.84–2.68 (m, 2H), 2.45–2.32 (m, 1H), 2.19–2.03 (m, 1H). 13C NMR (CDCl3) δ 190.2, 156.5, 150.2, 147.4, 139.0, 138.6, 135.7, 130.8, 129.9, 128.4 [determined by 1H–13C heteronuclear multiple-bond (HMBC) spectroscopy], 127.3, 126.8, 126.0, 122.6, 121.5, 112.9, 77.6, 42.6, 36.2, 31.2. Anal. (C20H15BrClNO2·0.3H2O) C, H, N.
N-(8-Bromo-6-chloro-4-oxo-2-phenethyl-4H-chromen-3-yl)acetamide (23)
Compound 22
was dissolved in pyridine (2 mL), and acetic anhydride (55 μL, 0.6
mmol) was added. The mixture was stirred at room temperature overnight.
The solution was coevaporated with toluene and EtOH. Purification by
flash chromatography EtOAc:heptane (4:6) gave 23 (0.19 g, 87%) as a white solid; mp 203 °C. 1H NMR (CDCl3) δ 8.09 (d, J = 2.6 Hz, 1H), 7.89 (d, J = 2.6 Hz, 1H), 7.32–7.17 (m, 5H), 6.91 (bs, 1H), 3.22–3.05 (m, 4H), 2.19 (s, 3H). 13C
NMR δ 189.2, 173.1, 169.3, 165.1, 151.0, 140.2, 136.9, 131.2, 128.7,
128.5, 126.6, 124.7, 124.3, 119.8, 112.8, 100.0, 34.1, 32.1, 23.6. Anal.
(C19H15BrClNO3) C, H, N.
(Z)-3-(Aminomethylene)-8-bromo-6-chloro-2-phenethylchoman-4-one (25)
A solution of 24 (0.025 g, 0.06 mmol) in anhydrous THF (2 mL) was cooled to −78 °C, and DIBAL-H (0.06 mL, 0.06 mmol, 1 M in CH2Cl2) was added dropwise to the mixture. After 1 h at −78 °C, a further portion of DIBAL-H (0.06 mL, 0.06 mmol, 1 M in CH2Cl2) was added. After 2 h, the reaction was quenched with saturated NH4Cl
(aq), followed by the addition of EtOAc. The aqueous phase was
extracted three times with EtOAc, and the combined organic phases were
washed with H2O and brine. The organic phase was dried over MgSO4 and concentrated under vacuum. Purification by flash chromatography using EtOAc:heptane (2:8 → 4:6) gave 25 (17 mg, 66%) as a beige solid; mp 106–108 °C. 1H NMR (CDCl3) δ 9.24 (br d, J = 10.3 Hz, 1H), 7.80 (d, J = 2.6 Hz, 1H), 7.61 (d, J = 2.6 Hz, 1H), 7.34–7.15 (m, 5H), 6.86–6.78 (m, 1H), 5.22 (br s, 1H), 4.86 (dd, J = 9.9, 4.4 Hz, 1H) 2.88–2.71 (m, 2H), 2.28–2.15 (m, 1H), 1.86–1.73 (m, 1H). 13C NMR (CDCl3)
δ 180.9, 153.3, 147.5, 141.0, 136.6, 128.74, 128.66, 126.9, 126.2,
125.7, 125.3, 112.5, 102.8, 79.2, 37.6, 31.8. The compound is 97% pure
according to HPLC analysis.
In Vitro Fluor de Lys Assay for SIRT1–3 Activities
The
Fluor de Lys fluorescence assays were based on the method described in
the BioMol product sheet (Enzo Life Sciences) using the BioMol KI177
substrate for SIRT1 and the KI179 substrate for SIRT2 and SIRT3. The
determined Km value of SIRT1 for KI177 was 58 μM, and the Km of SIRT2 for KI179 was 198 μM.(77) The Km of SIRT3 for KI179 was reported by BioMol to be 32 μM. The Km values of SIRT1, SIRT2, and SIRT3 for NAD+ were reported by BioMol to be 558 μM, 547 μM, and 2 mM, respectively.
Briefly, assays were carried out using the Fluor de Lys acetylated peptide substrate at a concentration corresponding to 0.7 Km and NAD+ (N6522, Sigma) at a concentration corresponding to 0.9 Km,
recombinant GST-SIRT1/2-enzyme or recombinant His-SIRT3 and SIRT assay
buffer (HDAC assay buffer, KI143, supplemented with 1 mg/mL BSA, A3803,
Sigma). GST-SIRT1 and GST-SIRT2 were produced as described previously.(78, 79) His-SIRT3 (BML-SE270) was purchased from Enzo Life Sciences. The buffer, Fluor de Lys acetylated peptide substrate, NAD+,
and DMSO/compounds in DMSO (2.5 μL in 50 μL total reaction volume; DMSO
from Sigma, D2650) were preincubated for 5 min at room temperature. The
reaction was started by adding the enzyme. The reaction mixture was
incubated for 1 h at 37 °C. After that, Fluor de Lys developer (KI176)
and nicotinamide (2 mM in HDAC assay buffer giving total volume of 50
μL) were added, and the incubation was continued for 45 min at 37 °C.
Fluorescence readings were obtained using EnVision 2104 multilabel
reader (PerkinElmer) with excitation wavelength 370 nm and emission 460
nm.
The IC50 values were
determined as three independent determinations with 8–10 different
inhibitor concentrations in each determination. This gave altogether 27
data points that were included in the calculation of the best-fit value
for nonlinear curve fitting with GraphPad Prism5 (GraphPad Software,
Inc.).
Histone Deacetylase (HDAC) Activity Assay
The
assay was done according to the instructions of the HDAC fluorimetric
assay/drug discovery kit (AK500, Enzo). Briefly, the assay was carried
out using 50 μM Fluor de Lys HDAC substrate (KI104, Enzo) and 200 μM
test compounds with HeLa cell nuclear extract (KI410, Enzo) as a source
of HDAC enzymatic activity. The reaction mixture was incubated for 1 h
at 37 °C. After that, Fluor de Lys developer plus 1 μM Trichostatin A
was added and incubation was continued for 15 min at 25 °C. Fluorescence
readings were obtained with EnVision 2104 multilabel reader
(PerkinElmer) with excitation wavelength 370 nm and emission 460 nm.
SIRTainty Sirtuin Activity Assay
The
fluorometric SIRTainty class III HDAC assay (Millipore) employs
nicotinamidase to measure nicotinamide generated upon the cleavage of
NAD+ during sirtuin mediated deacetylation of a substrate.(80) The SIRT2 activity testing was performed according to the assay instructions in the presence and absence of NAD+.
The results were read using Victor 1420 multilabel counter (Wallac)
with excitation wavelength 405 nm and emission 460 nm and were reported
as % of inhibition of the NAD+-dependent signal.
Cell Culture
Human
A549 lung carcinoma cells and MCF-7 breast carcinoma cells (both from
ATCC) were maintained in Dulbecco’s Modified Eagle Medium (DMEM)
containing 10% fetal calf serum, 100 U/mL penicillin, and 100 μg/mL
streptomycin (all from Gibco) at +37 °C in a humidified atmosphere of 5%
CO2/95% air.
Cell Proliferation and Cell Cycle Analysis
For
cell proliferation assays with sulforhodamine B, the cells were plated
to 96-well plates (Nunc) 24 h before the start of the treatments (3000
cells/well). The cells were treated with vehicle (0.5% DMSO) or test
compounds for 48 h (A549 cells) or 72 h (MCF-7 cells). Sulforhodamine B
staining was performed as previously described.(64)
For cell cycle analysis with propidium iodide staining, the cells were
plated to 6-well plates (Nunc) 6 h before the start of the treatments
(0.6 × 106 cells/well). The cells were treated with vehicle
(0.5% DMSO) or test compounds for 18 h. Propidium iodide staining was
performed as previously described.(64) FACScanto II flow cytometer with FACSDiva software (Becton Dickinson) was used to analyze cellular DNA content and cell cycle.
Western Blotting
The MCF-7 cells were plated to 12-well plates (Nunc) at a density of 105
cells/well, and the experiments were initiated after 24 h. The cells
were treated with vehicle (0.5% DMSO) or test compounds as previously
described.(81)
For the analysis of α-tubulin acetylation levels, the cells were lysed
into M-PER mammalian protein extraction reagent (Thermo Fisher
Scientific) followed by centrifugation (20 min, 16000g, +4 °C).
After electrophoretic separation in SDS-PAGE gel, the proteins were
transferred onto Hybond-ECL nitrocellulose transfer membrane (GE
Healthcare) and were detected with mouse monoclonal acetylated α-tubulin
antibody (T6793, Sigma) and total α-tubulin antibody (T5168, Sigma).
The protein signals were visualized with peroxidase-conjugated sheep
antimouse secondary antibody (ab97046, Abcam) and ECL Plus
chemiluminescent substrate (GE Healthcare). The images were obtained by
the use of digital imaging (ImageQuant, GE Healthcare).
Statistical Analyses
Statistical
differences between groups were tested using one-way analysis of
variance (ANOVA), followed by Tukey′s Multiple Comparison Test, with p
< 0.05 considered as statistically significant. Data analysis was
performed using GraphPad Prism version 5.03 for Windows (GraphPad
Software).
Supporting Information
Characterization
data and elemental analysis or HRMS analyses for all tested compounds;
complete list of SIRT1–3 activity assay results; list of calculated
physicochemical properties of all tested compounds; description of the
SIRT2 homology modeling procedure and validation; 1H NMR and 13C NMR spectra of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Acknowledgment
We
thank the Swedish Research Council (project no. 621-2013-4749), the
Academy of Finland (grant nos. 127062 and 132780), and the Department of
Chemistry and Molecular Biology, University of Gothenburg, for
financial support and Biocenter Kuopio for providing facilities. We also
thank Johanna Stéen for skillful assistance in the lab. This work is
part of COST Action TD09056: “Epigenetics: Bench to Bedside”.
| Abbreviation Used | |
| Ac |
acetyl
|
| ADP |
adenosine diphosphate
|
| ADPr |
adenosine diphosphate ribose
|
| ANOVA |
analysis of variance
|
| BSA |
bovine serum albumin
|
| DDQ |
2,3-dichloro-5,6-dicyano-p-benzoquinone
|
| DIBAL-H |
diisobutylaluminum hydride
|
| DIPA |
N,N-diisopropylamine
|
| DMF |
dimethylformamide
|
| DMSO |
dimethyl sulfoxide
|
| DNA |
deoxyribnucleic acid
|
| ECL |
enhanced chemiluminescence
|
| ESI-MS |
electrospray ionization mass spectrometry
|
| FOX |
Forkhead box protein
|
| GST |
glutathione-S-transferase
|
| HDAC |
histone deacetylase
|
| HPLC |
high-performance liquid chromatography
|
| IC50 |
the half-maximal inhibitory concentration
|
| KHMDS |
potassium bis(trimethylsilyl)amide
|
| LDA |
lithium diisopropylamide
|
| LDS |
lithium dodecyl sulfate
|
| Ms |
methanesulfonyl
|
| MW |
microwave
|
| NAD+ |
nicotinamide adenine dinucleotide
|
| NMR |
nuclear magnetic resonance
|
| PDB |
Protein Data Bank
|
| SAR |
structure–activity relationship
|
| SDS-PAGE |
sodium dodecyl sulfate polyacrylamide gel electrophoresis
|
| SEM |
standard error of the mean
|
| SIRT |
silent information regulator type
|
| TBAA |
tetra-N-butylammonium acetate
|
| TBDMS |
tert-butyldimethylsilyl
|
| TEMPO |
(2,2,6,6-tetramethylpiperidin-1-yl)oxy
|
| THF |
tetrahydrofuran
|
| TLC |
thin-layer chromatography
|
| TPAP |
tetrapropylammonium perruthenate
|
| Ts |
tosyl
|
| p-TSA |
p-toluenesulfonic acid
|
| UV |
ultraviolet
|
References
This article references 81 other publications.
- 4.Sauve, A. A.; Celic, I.; Avalos, J.; Deng, H. T.; Boeke, J. D.; Schramm, V. L. Chemistry of gene silencing: the mechanism of NAD(+)-dependent deacetylation reactions Biochemistry 2001, 40, 15456– 15463[ACS Full Text
 ], [CAS]
], [CAS] - 10.Du, J. T.; Zhou, Y. Y.; Su, X. Y.; Yu, J. J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J. H.; Choi, B. H.; He, B.; Chen, W.; Zhang, S.; Cerione, R. A.; Auwerx, J.; Hao, Q.; Lin, H. N. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase Science 2011, 334, 806– 809
- 11.Tan, M. J.; Peng, C.; Anderson, K. A.; Chhoy, P.; Xie, Z. Y.; Dai, L. Z.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; Ro, J.; Wagner, G. R.; Green, M. F.; Madsen, A. S.; Schmiesing, J.; Peterson, B. S.; Xu, G. F.; Ilkayeva, O. R.; Muehlbauer, M. J.; Braulke, T.; Muhlhausen, C.; Backos, D. S.; Olsen, C. A.; McGuire, P. J.; Pletcher, S. D.; Lombard, D. B.; Hirschey, M. D.; Zhao, Y. M. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5 Cell Metab. 2014, 19, 605– 617
- 15.Smith, M. R.; Syed, A.; Lukacsovich, T.; Purcell, J.; Barbaro, B. A.; Worthge, S. A.; Wei, S. R.; Pollio, G.; Magnoni, L.; Scali, C.; Massai, L.; Franceschini, D.; Camarri, M.; Gianfriddo, M.; Diodato, E.; Thomas, R.; Gokce, O.; Tabrizi, S. J.; Caricasole, A.; Landwehrmeyer, B.; Menalled, L.; Murphy, C.; Ramboz, S.; Luthi-Carter, R.; Westerberg, G.; Marsh, J. L. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease Hum. Mol. Genet. 2014, 23, 2995– 3007
- 16.A Phase II Safety and Tolerability Study With SEN0014196. ClinicalTrials.gov; U.S. National Library of Medicine, U.S. National Institutes of Health: Bethesda, MD, 2012; NCT01521585, https://clinicaltrials.gov/ct2/show/NCT01521585?term=nct01521585&rank=1.
- 20.Taylor, D. M.; Balabadra, U.; Xiang, Z. M.; Woodman, B.; Meade, S.; Amore, A.; Maxwell, M. M.; Reeves, S.; Bates, G. P.; Luthi-Carter, R.; Lowden, P. A. S.; Kazantsev, A. G. A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase ACS Chem. Biol. 2011, 6, 540– 546[ACS Full Text], [CAS]
- 21.Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K. L.; Edgerly, C.; Cipicchio, P. M.; Lauver, M. A.; Choi, S. H.; Silverman, R. B.; Ferrante, R. J.; Hersch, S.; Kazantsev, A. G. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models Cell Rep. 2012, 2, 1492– 1497
- 22.Outeiro, T. F.; Kontopoulos, E.; Altmann, S. M.; Kufareva, I.; Strathearn, K. E.; Amore, A. M.; Volk, C. B.; Maxwell, M. M.; Rochet, J. C.; McLean, P. J.; Young, A. B.; Abagyan, R.; Feany, M. B.; Hyman, B. T.; Kazantsev, A. G. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease Science 2007, 317, 516– 519
- 27.Li, Y. Z.; Matsumori, H.; Nakayama, Y.; Osaki, M.; Kojima, H.; Kurimasa, A.; Ito, H.; Mori, S.; Katoh, M.; Oshimura, M.; Inoue, T. SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis Genes Cells 2011, 16, 34– 45
- 29.Liu, P. Y.; Xu, N.; Malyukova, A.; Scarlett, C. J.; Sun, Y. T.; Zhang, X. D.; Ling, D.; Su, S. P.; Nelson, C.; Chang, D. K.; Koach, J.; Tee, A. E.; Haber, M.; Norris, M. D.; Toon, C.; Rooman, I.; Xue, C.; Cheung, B. B.; Kumar, S.; Marshall, G. M.; Biankin, A. V.; Liu, T. The histone deacetylase SIRT2 stabilizes Myc oncoproteins Cell Death Differ. 2013, 20, 503– 514
- 32.Fridén-Saxin, M.; Seifert, T.; Landergren, M. R.; Suuronen, T.; Lahtela-Kakkonen, M.; Jarho, E. M.; Luthman, K. Synthesis and evaluation of substituted chroman-4-one and chromone derivatives as sirtuin 2-selective inhibitors J. Med. Chem. 2012, 55, 7104– 7113[ACS Full Text], [CAS]
- 33.McDougal, P. G.; Rico, J. G.; Oh, Y. I.; Condon, B. D. A Convenient procedure for the monosilylation of symmetrical 1,n-diols J. Org. Chem. 1986, 51, 3388– 3390[ACS Full Text], [CAS]
- 34.Shah, S. T. A.; Singh, S.; Guiry, P. J. A Novel, Chemoselective and efficient microwave-assisted deprotection of silyl ethers with Selectfluor J. Org. Chem. 2009, 74, 2179– 2182[ACS Full Text], [CAS]
- 37.Zaragoza, F.; Stephensen, H.; Peschke, B.; Rimvall, K. 2-(4-Alkylpiperazin-1-yl)quinolines as a new class of imidazole-free histamine H-3 receptor antagonists J. Med. Chem. 2005, 48, 306– 311[ACS Full Text], [CAS]
- 38.Battistuzzi, G.; Cacchi, S.; Fabrizi, G. An efficient palladium-catalyzed synthesis of cinnamaldehydes from acrolein diethyl acetal and aryl iodides and bromides Org. Lett. 2003, 5, 777– 780[ACS Full Text], [CAS]
- 40.Fridén-Saxin, M.; Pemberton, N.; Andersson, K. D.; Dyrager, C.; Friberg, A.; Grøtli, M.; Luthman, K. Synthesis of 2-alkyl-substituted chromone derivatives using microwave irradiation J. Org. Chem. 2009, 74, 2755– 2759[ACS Full Text], [CAS]
- 41.Ankner, T.; Friden-Saxin, M.; Pemberton, N.; Seifert, T.; Grøtli, M.; Luthman, K.; Hilmersson, G. KHMDS enhanced SmI2-mediated Reformatsky type α-cyanation Org. Lett. 2010, 12, 2210– 2213[ACS Full Text], [CAS]
- 44.Borra, M. T.; Langer, M. R.; Slama, J. T.; Denu, J. M. Substrate specificity and kinetic mechanism of the Sir2 family of NAD(+)-dependent histone/protein deacetylases Biochemistry 2004, 43, 9877– 9887[ACS Full Text], [CAS]
- 45.Jin, L.; Wei, W. T.; Jiang, Y. B.; Peng, H.; Cai, J. H.; Mao, C.; Dai, H.; Choy, W.; Bemis, J. E.; Jirousek, M. R.; Milne, J. C.; Westphal, C. H.; Perni, R. B. Crystal structures of human SIRT3 displaying substrate-induced conformational changes J. Biol. Chem. 2009, 284, 24394– 24405
- 47.Disch, J. S.; Evindar, G.; Chiu, C. H.; Blum, C. A.; Dai, H.; Jin, L.; Schuman, E.; Lind, K. E.; Belyanskaya, S. L.; Deng, J. H.; Coppo, F.; Aquilani, L.; Graybill, T. L.; Cuozzo, J. W.; Lavu, S.; Mao, C.; Vlasuk, G. P.; Perni, R. B. Discovery of thieno[3,2-4-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3 J. Med. Chem. 2013, 56, 3666– 3679[ACS Full Text], [CAS]
- 48.Huber, K.; Schemies, J.; Uciechowska, U.; Wagner, J. M.; Rumpf, T.; Lewrick, F.; Suss, R.; Sippl, W.; Jung, M.; Bracher, F. Novel 3-arylideneindolin-2-ones as inhibitors of NAD(+)-dependent histone deacetylases (Sirtuins) J. Med. Chem. 2010, 53, 1383– 1386[ACS Full Text], [CAS]
- 49.Rotili, D.; Tarantino, D.; Nebbioso, A.; Paolini, C.; Huidobro, C.; Lara, E.; Mellini, P.; Lenoci, A.; Pezzi, R.; Botta, G.; Lahtela-Kakkonen, M.; Poso, A.; Steinkuhler, C.; Gallinari, P.; De Maria, R.; Fraga, M.; Esteller, M.; Altucci, L.; Mai, A. Discovery of salermide-related sirtuin inhibitors: binding mode studies and antiproliferative effects in cancer cells including cancer stem cells J. Med. Chem. 2012, 55, 10937– 10947[ACS Full Text], [CAS]
- 50.Medda, F.; Russell, R. J. M.; Higgins, M.; McCarthy, A. R.; Campbell, J.; Slawin, A. M. Z.; Lane, D. P.; Lain, S.; Westwood, N. J. Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity J. Med. Chem. 2009, 52, 2673– 2682[ACS Full Text], [CAS]
- 51.Neugebauer, R. C.; Uchiechowska, U.; Meier, R.; Hruby, H.; Valkov, V.; Verdin, E.; Sippl, W.; Jung, M. Structure–activity studies on splitomicin derivatives as sirtuin inhibitors and computational prediction of binding mode J. Med. Chem. 2008, 51, 1203– 1213[ACS Full Text], [CAS]
- 55.Yamagata, K.; Goto, Y.; Nishimasu, H.; Morimoto, J.; Ishitani, R.; Dohmae, N.; Takeda, N.; Nagai, R.; Komuro, I.; Suga, H.; Nureki, O. Structural basis for potent inhibition of SIRT2 deacetylase by a macrocyclic peptide inducing dynamic structural change Structure 2014, 22, 345– 352
- 56.Zhao, X.; Allison, D.; Condon, B.; Zhang, F. Y.; Gheyi, T.; Zhang, A. P.; Ashok, S.; Russell, M.; MacEwan, I.; Qian, Y. W.; Jamison, J. A.; Luz, J. G. The 2.5 Å crystals of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD(+)) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition J. Med. Chem. 2013, 56, 963– 969[ACS Full Text], [CAS]
- 59.Wilcken, R.; Zimmermann, M. O.; Lange, A.; Joerger, A. C.; Boeckler, F. M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology J. Med. Chem. 2013, 56, 1363– 1388[ACS Full Text], [CAS]
- 62.Friesner, R. A.; Murphy, R. B.; Repasky, M. P.; Frye, L. L.; Greenwood, J. R.; Halgren, T. A.; Sanschagrin, P. C.; Mainz, D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes J. Med. Chem. 2006, 49, 6177– 6196[ACS Full Text], [CAS]
- 63.The docking was performed using the Glide docking tool as implemented in the Schrödinger software.
- 64.Mellini, P.; Kokkola, T.; Suuronen, T.; Salo, H. S.; Tolvanen, L.; Mai, A.; Lahtela-Kakkonen, M.; Jarho, E. M. Screen of pseudopeptidic inhibitors of human Sirtuins 1–3: two lead compounds with antiproliferative effects in cancer cells J. Med. Chem. 2013, 56, 6681– 6695[ACS Full Text], [CAS]
- 65.Smith, B. C.; Denu, J. M. Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide Biochemistry 2007, 46, 14478– 14486[ACS Full Text], [CAS]
- 68.McCarthy, A. R.; Sachweh, M. C. C.; Higgins, M.; Campbell, J.; Drummond, C. J.; van Leeuwen, I. M. M.; Pirrie, L.; Ladds, M. J. G. W.; Westwood, N. J.; Lain, S. Tenovin-D3, a novel small-molecule inhibitor of sirtuin SirT2, increases p21 (CDKN1A) expression in a p53-independent manner Mol. Cancer Ther. 2013, 12, 352– 360
- 71.Taillier, C.; Gille, B.; Bellosta, V.; Cossy, J. Synthetic approaches and total synthesis of natural zoapatanol J. Org. Chem. 2005, 70, 2097– 2108[ACS Full Text], [CAS]
- 72.Frankowski, K. J.; Golden, J. E.; Zeng, Y. B.; Lei, Y.; Aube, J. Syntheses of the stemona alkaloids (±)-stenine, (±)-neostenine, and (±)-13-epineostenine using a stereodivergent Diels–Alder/azido-Schmidt reaction J. Am. Chem. Soc. 2008, 130, 6018– 6024[ACS Full Text], [CAS]
- 73.Kaiser, F.; Schwink, L.; Velder, J.; Schmalz, H. G. Studies toward the total synthesis of mumbaistatin, a highly potent glucose-6-phosphate translocase inhibitor. Synthesis of a mumbaistatin analogue J. Org. Chem. 2002, 67, 9248– 9256[ACS Full Text], [CAS]
- 74.Kitbunnadaj, R.; Zuiderveld, O. P.; Christophe, B.; Hulscher, S.; Menge, W. M. P. B.; Gelens, E.; Snip, E.; Bakker, R. A.; Celanire, S.; Gillard, M.; Talaga, P.; Timmerman, H.; Leurs, R. Identification of 4-(1H-imidazol-4(5)-ylmethyl)pyridine (immethridine) as a novel, potent, and highly selective histamine H-3 receptor agonist J. Med. Chem. 2004, 47, 2414– 2417[ACS Full Text], [CAS]
- 77.Kiviranta, P. H.; Suuronen, T.; Wallén, E. A. A.; Leppänen, J.; Tervonen, J.; Kyrylenko, S.; Salminen, A.; Poso, A.; Jarho, E. M. N-epsilon-Thioacetyl-lysine-containing tri-, tetra-, and pentapeptides as SIRT1 and SIRT2 inhibitors J. Med. Chem. 2009, 52, 2153– 2156[ACS Full Text], [CAS]
- 78.Kiviranta, P. H.; Leppänen, J.; Rinne, V. M.; Suuronen, T.; Kyrylenko, O.; Kyrylenko, S.; Kuusisto, E.; Tervo, A. J.; Järvinen, T.; Salminen, A.; Poso, A.; Wallén, E. A. A. N-(3-(4-Hydroxyphenyl)-propenoyl)-amino acid tryptamides as SIRT2 inhibitors Bioorg. Med. Chem. Lett. 2007, 17, 2448– 2451
- 79.Tervo, A. J.; Kyrylenko, S.; Niskanen, P.; Salminen, A.; Leppänen, J.; Nyronen, T. H.; Järvinen, T.; Poso, A. An in silico approach to discovering novel inhibitors of human sirtuin type 2 J. Med. Chem. 2004, 47, 6292– 6298[ACS Full Text], [CAS]
- 80.Hubbard, B. P.; Gomes, A. P.; Dai, H.; Li, J.; Case, A. W.; Considine, T.; Riera, T. V.; Lee, J. E.; Yen, E. S.; Lamming, D. W.; Pentelute, B. L.; Schuman, E. R.; Stevens, L. A.; Ling, A. J. Y.; Armour, S. M.; Michan, S.; Zhao, H. Z.; Jiang, Y.; Sweitzer, S. M.; Blum, C. A.; Disch, J. S.; Ng, P. Y.; Howitz, K. T.; Rolo, A. P.; Hamuro, Y.; Moss, J.; Perni, R. B.; Ellis, J. L.; Vlasuk, G. P.; Sinclair, D. A. Evidence for a common mechanism of SIRT1 regulation by allosteric activators Science 2013, 339, 1216– 1219
- 81.Heltweg, B.; Gatbonton, T.; Schuler, A. D.; Posakony, J.; Li, H. Z.; Goehle, S.; Kollipara, R.; DePinho, R. A.; Gu, Y. S.; Simon, J. A.; Bedalov, A. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes Cancer Res. 2006, 66, 4368– 4377
Citation data is made available by participants in
Crossref's
Cited-by Linking
service. For a more comprehensive list of citations to this article, users are
encouraged to perform a search inSciFinder.
 Nadja A. SimethLisa-Marie AltmannNathalie WössnerElisabeth BauerManfred JungBurkhard KönigThe Journal of Organic Chemistry 2018 83 (15), 7919-7927
Nadja A. SimethLisa-Marie AltmannNathalie WössnerElisabeth BauerManfred JungBurkhard KönigThe Journal of Organic Chemistry 2018 83 (15), 7919-7927 Matthias Schiedel, Daniel Herp, Sören Hammelmann, Sören Swyter, Attila Lehotzky, Dina Robaa, Judit Oláh, Judit Ovádi, Wolfgang Sippl, and Manfred JungJournal of Medicinal Chemistry 2018 61 (2), 482-491
Matthias Schiedel, Daniel Herp, Sören Hammelmann, Sören Swyter, Attila Lehotzky, Dina Robaa, Judit Oláh, Judit Ovádi, Wolfgang Sippl, and Manfred JungJournal of Medicinal Chemistry 2018 61 (2), 482-491 Joana Reis, Alexandra Gaspar, Nuno Milhazes, and Fernanda BorgesJournal of Medicinal Chemistry 2017 60 (19), 7941-7957
Joana Reis, Alexandra Gaspar, Nuno Milhazes, and Fernanda BorgesJournal of Medicinal Chemistry 2017 60 (19), 7941-7957 Shenzhen Huang, Chunli Song, Xiang Wang, Guo Zhang, Yanlin Wang, Xiaojuan Jiang, Qizheng Sun, Luyi Huang, Rong Xiang, Yiguo Hu, Linli Li, and Shengyong YangJournal of Chemical Information and Modeling 2017 57 (4), 669-679
Shenzhen Huang, Chunli Song, Xiang Wang, Guo Zhang, Yanlin Wang, Xiaojuan Jiang, Qizheng Sun, Luyi Huang, Rong Xiang, Yiguo Hu, Linli Li, and Shengyong YangJournal of Chemical Information and Modeling 2017 57 (4), 669-679 Sébastien Moniot, Mariantonietta Forgione, Alessia Lucidi, Gebremedhin S. Hailu, Angela Nebbioso, Vincenzo Carafa, Francesca Baratta, Lucia Altucci, Nicola Giacché, Daniela Passeri, Roberto Pellicciari, Antonello Mai, Clemens Steegborn, and Dante RotiliJournal of Medicinal Chemistry 2017 60 (6), 2344-2360
Sébastien Moniot, Mariantonietta Forgione, Alessia Lucidi, Gebremedhin S. Hailu, Angela Nebbioso, Vincenzo Carafa, Francesca Baratta, Lucia Altucci, Nicola Giacché, Daniela Passeri, Roberto Pellicciari, Antonello Mai, Clemens Steegborn, and Dante RotiliJournal of Medicinal Chemistry 2017 60 (6), 2344-2360 Sandeep Sundriyal, Sébastien Moniot, Zimam Mahmud, Shang Yao, Paolo Di Fruscia, Christopher R. Reynolds, David T. Dexter, Michael J. E. Sternberg, Eric W.-F. Lam, Clemens Steegborn, and Matthew J. FuchterJournal of Medicinal Chemistry 2017 60 (5), 1928-1945
Sandeep Sundriyal, Sébastien Moniot, Zimam Mahmud, Shang Yao, Paolo Di Fruscia, Christopher R. Reynolds, David T. Dexter, Michael J. E. Sternberg, Eric W.-F. Lam, Clemens Steegborn, and Matthew J. FuchterJournal of Medicinal Chemistry 2017 60 (5), 1928-1945- Tina Seifert, Marcus Malo, Johan Lengqvist, Carina Sihlbom, Elina M. Jarho, and Kristina LuthmanJournal of Medicinal Chemistry 2016 59 (23), 10794-10799
- Azim Ziyaei Halimehjani and Maryam KhoshdounThe Journal of Organic Chemistry 2016 81 (13), 5699-5704
- Haruhisa Kikuchi, Tsuyoshi Hoshikawa, Shoichiro Kurata, Yasuhiro Katou, and Yoshiteru OshimaJournal of Natural Products 2016 79 (5), 1259-1266
X-ray crystal structure guided discovery of new selective, substrate-mimicking sirtuin 2 inhibitors that exhibit activities against non-small cell lung cancer cells
Ling-Ling Yang, Hua-Li Wang, Lei Zhong, Chen Yuan, Si-Yu Liu, Zhu-Jun Yu, Sha Liu, Yu-Hang Yan, Chengyong Wu, Yuxi Wang, Zhouyu Wang, Yamei Yu, Qiang Chen, Guo-Bo LiEuropean Journal of Medicinal Chemistry 2018 155, 806-823New chemical tools for probing activity and inhibition of the NAD + -dependent lysine deacylase sirtuin 2
Sören Swyter, Matthias Schiedel, Daria Monaldi, Sándor Szunyogh, Attila Lehotzky, Tobias Rumpf, Judit Ovádi, Wolfgang Sippl, Manfred JungPhilosophical Transactions of the Royal Society B: Biological Sciences 2018 373 (1748), 20170083Novel class of mononuclear 2-methoxy-4-chromanones ligated Cu (II), Zn (II), Ni (II) complexes: synthesis, characterisation and biological studies
E. Sneha Jose, Jessica Elizabeth Philip, A.A. Shanty, M.R.P. Kurup, P.V. MohananInorganica Chimica Acta 2018 478, 155-165The Current State of NAD + -Dependent Histone Deacetylases (Sirtuins) as Novel Therapeutic Targets
Matthias Schiedel, Dina Robaa, Tobias Rumpf, Wolfgang Sippl, Manfred JungMedicinal Research Reviews 2018 38 (1), 147-200Metal-free oxidative decarbonylative alkylation of chromones using aliphatic aldehydes
Rongzhen Chen, Jin-Tao Yu, Jiang ChengOrganic & Biomolecular Chemistry 2018 16 (19), 3568-3571Transition-metal-free direct C-3 alkylation of quinoxalin-2(1 H )-ones with ethers
Jinwei Yuan, Junhao Fu, Jihong Yin, Zhenhua Dong, Yongmei Xiao, Pu Mao, Lingbo QuOrganic Chemistry Frontiers 2018 14,Silver-catalyzed decarboxylative cascade radical cyclization of tert -carboxylic acids and o -(allyloxy)arylaldehydes towards chroman-4-one derivatives
Hao Hu, Xiaolan Chen, Kai Sun, Junchao Wang, Yan Liu, Hui Liu, Bing Yu, Yuanqiang Sun, Lingbo Qu, Yufen ZhaoOrganic Chemistry Frontiers 2018 117,Synthesis, anticancer, structural, and computational docking studies of 3-benzylchroman-4-one derivatives
Lalitha Simon, Abdul Ajees Abdul Salam, S. Madan Kumar, T. Shilpa, K.K. Srinivasan, K. ByrappaBioorganic & Medicinal Chemistry Letters 2017 27 (23), 5284-5290Visible Light-Promoted Synthesis of Spiroepoxy Chromanone Derivatives via a Tandem Oxidation/Radical Cyclization/Epoxidation Process
Sungwoo Jung, Jiyun Kim, Sungwoo HongAdvanced Synthesis & Catalysis 2017 359 (22), 3945-3949New chroman-4-one/thiochroman-4-one derivatives as potential anticancer agents
Seref Demirayak, Leyla Yurttas, Nalan Gundogdu-Karaburun, Ahmet Cagri Karaburun, Ismail KayagilSaudi Pharmaceutical Journal 2017 25 (7), 1063-1072Aldehydes as Carbon Radical Acceptors: Silver Nitrate Catalyzed Cascade Decarboxylation and Oxidative Cyclization toward Dihydroflavonoid Derivatives
Wen-Chao Yang, Peng Dai, Kai Luo, Yi-Gang Ji, Lei WuAdvanced Synthesis & Catalysis 2017 359 (14), 2390-2395A facile one-pot regioselective synthesis of functionalized novel benzo[f]chromeno[4,3-b][1,7]naphthyridines and benzo[f][1,7]naphthyridines via an imino Diels-Alder reaction
Sateesh Kumar Arepalli, Byeongwoo Park, Jae-Kyung Jung, Kiho Lee, Heesoon LeeTetrahedron Letters 2017 58 (5), 449-454A highly enantioselective access to chiral chromanones and thiochromanones via copper-catalyzed asymmetric conjugated reduction of chromones and thiochromones
Donglu Xiong, Wenxi Zhou, Zhiwu Lu, Suping Zeng, Jun (Joelle) WangChemical Communications 2017 53 (51), 6844-6847Unprecedented formation of 3-(tetrahydrofuran-2-yl)-4H-chromen-4-one in a reaction between 3,3a-dihydro-9H-furo[3,4-b]chromen-9-one and malononitrile
Jie-Jie Liu, Liang Cheng, Hong-Yan Huang, Feng Wei, Dong Wang, Li LiuOrganic & Biomolecular Chemistry 2017 15 (23), 5078-5088Sirtuin functions and modulation: from chemistry to the clinic
Vincenzo Carafa, Dante Rotili, Mariantonietta Forgione, Francesca Cuomo, Enrica Serretiello, Gebremedhin Solomon Hailu, Elina Jarho, Maija Lahtela-Kakkonen, Antonello Mai, Lucia AltucciClinical Epigenetics 2016 8 (1),Role of Sirtuins in Maintenance of Genomic Stability: Relevance to Cancer and Healthy Aging
Xiayu Wu, Neng Cao, Michael Fenech, Xu WangDNA and Cell Biology 2016 35 (10), 542-5755-Benzylidene-hydantoin is a new scaffold for SIRT inhibition: From virtual screening to activity assays
Lionel Sacconnay, Lucie Ryckewaert, Giuseppe Marco Randazzo, Charlotte Petit, Carolina Dos Santos Passos, Jelena Jachno, Vilma Michailovienė, Asta Zubrienė, Daumantas Matulis, Pierre-Alain Carrupt, Claudia Avello Simões-Pires, Alessandra NurissoEuropean Journal of Pharmaceutical Sciences 2016 85, 59-67A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity
Hui Jing, Jing Hu, Bin He, Yashira L. Negrón Abril, Jack Stupinski, Keren Weiser, Marisa Carbonaro, Ying-Ling Chiang, Teresa Southard, Paraskevi Giannakakou, Robert S. Weiss, Hening LinCancer Cell 2016 29 (3), 297-310Isocyanide and Meldrum's acid-based multicomponent reactions in diversity-oriented synthesis: from a serendipitous discovery towards valuable synthetic approaches
Ahmad Shaabani, Seyyed Emad HooshmandRSC Advances 2016 6 (63), 58142-58159Straightforward synthesis of functionalized chroman-4-ones through cascade radical cyclization-coupling of 2-(allyloxy)arylaldehydes
Jingjing Zhao, Pan Li, Xinjian Li, Chungu Xia, Fuwei LiChemical Communications 2016 52 (18), 3661-3664Recent advances of chroman-4-one derivatives: Synthetic approaches and bioactivities
Saeed Emami, Zahra GhanbarimasirEuropean Journal of Medicinal Chemistry 2015 93, 539-563





- Retrieve Detailed Record of this Article
- Retrieve Substances Indexed for this Article
- Retrieve Reactions Indexed for this Article
- Retrieve All References Cited for this Article
- Retrieve All References Citing this Article
Metrics 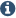
+
 More Article Metrics
More Article Metrics
- COVER STORY
Puerto Rico se levanta
When Hurricane Maria swept across Puerto Rico last year, no one was prepared for its destruction. Though... - SCIENCE CONCENTRATES

New CRISPR inhibitors found
As scientists march closer to using CRISPR gene editing to treat diseases, the U.S. Department of Defense... - BUSINESS CONCENTRATES
Broad prevails over Berkeley in CRISPR patent dispute
A long legal dispute over the rights to CRISPR/Cas9 gene editing has climaxed. In a decision released... - POLICY CONCENTRATES
EPA toxicity analysis of GenX-related compound coming soon
The U.S. Environmental Protection Agency will soon release a toxicity assessment for hexafluoropropylene...
Chemists predict ghostly molecule
Electromagnetic fields could create molecule-like electron density around single atom, according to computer simulationsSABIC, Clariant start to work together
Following an investment, the firms decide to combine two polymer businessesBile acids sneak nanoparticles into the bloodstream
Researchers fed rats nanoparticles coated in bile acids in a step toward to making ...Novo Nordisk R&D restructuring signals move to finding innovation externally
The drug company is cutting 400 R&D positions and opening 4 new research centers ...Perkin Medal winner reflects on her role in the science of keeping cool
Chemical engineer Barbara Minor talks about a career that advanced environmentally safer refrigerants
Read Impactful Research Highlights from Langmuir
Langmuir is the leading journal focusing on the fundamental science of systems and materials ...Read High-Impact Research Highlights from Biochemistry
Biochemistry provides an international forum for publishing exceptional, rigorous, high-impact research across all of ...Read High-Impact Research from The Journal of Organic Chemistry
The Journal of Organic Chemistry (JOC) welcomes original contributions of fundamental research in all branches ...Read High-Impact Research from Inorganic Chemistry
Inorganic Chemistry publishes fundamental studies in all phases of inorganic chemistry and has earned respect ...Read Impactful Research Highlights from ACS Chemical Neuroscience
ACS Chemical Neuroscience publishes high-quality research articles and reviews that showcase chemical, quantitative biological, ...Free to Read, Free to Publish: High-Impact Highlights from ACS Central Science
ACS Central Science, the Society’s award-winning fully open access journal, is fast rising to ...
---
2014- 2016 HUOM tästä löytyy myös abstraktin 2014 jälkeistä tutkimustulosta vuodelta 2016
Inga kommentarer:
Skicka en kommentar